Переведення проби у розчин
Сучасні методи підготовки проби базуються на використанні високо активних реагентів, збільшених тиску та температури, каталізу, випромінювань різного типу. Вибір способу розкладання проби та переведення її компонентів у розчин залежать від деяких факторів, які необхідно враховувати під час обгрунтування схеми хімічного аналізу. Перед усім звертають увагу на неорганічну чи органічну природу основи об’єкту (матріца), хімічний склад зразку, хімічні властивості компонента, що визначають.
Спосіб розкладання проби та переведення її у розчин залежить від мети аналізу. Так, різним чином проводять пробопідготовку під час елементного та функціонального аналізу органічних сполук; під час визначення загального вмісту будь-якого елементу (залізо, хром) та його форм у різному ступені окиснення (ферум(ІІ) та ферум(ІІІ), хром(ІІІ) та хром(VI)), а також основних компонентів зразку та домішок у ньому.
Спосіб розкладання та переведення проби в багатьом визначається вибраним аналітичним методом. Наприклад, пробопідготовка під час визначення органічних сполук у біологічних об’єктах хроматографічним та спектрофотометричними методами розрізняється.
Після вибору способу розкладання проби необхідно оцінити усі можливі похибки на цій стадії аналізу. Найбільш типові похибки обумовлені втратами летучих компонентів під час використання високих температур; забрудненням із матеріалів посуду та пристріїв для розкладання проб; наявністю заважаючих проведенню аналізу домішок у реактивах та розчинниках, що використовують для розкладання зразків.
Способи розкладання поділяють на “сухі” та “мокрі”: до перших відносять термічне розкладання, сплавлення та зпікання з різними речовинами (солі, оксиди, луги та їх суміші); до других ¾ розчинення досліджуваної проби у різних розчинниках, переважно у кислотах та сумішах.
Розчинення
У найкращому випадку розчинник повинен розчиняти пробу швидко, у достатньо м’яких умовах і не заважати на слідуючих стадіях аналізу.
Найкращий розчинник ¾ вода. Багато неорганічних солей та деякі органічні сполуки (нижчі та багатоатомні спирти, амінокислоти, гідрохлориди амінів, солі лужних металів органічних кислот, сечовина) легко розчиняються у воді. Іноді у воду додають невелику кількість кислоти для запобігання гідролізу та часткового осадження деяких катіонів металів. У окремих випадках для розчинення органічних речовин використовують суміш води та органічного розчинника, що змішується з нею (наприклад, суміш води та етанолу).
Для розчинення органічних сполук застосовують органічні розчинники. Як правило, це спирти, хлоровані вуглеводні, кетони. Так, для розчинення полімерних матеріалів різного типу використовують диметилформамід, диметил ацетамід, метилізобутилкетон, метанол.
Під час “мокрого” способу розкладання проби часто застосовують різні кислоти та їх суміші за умов нагрівання. При цьому в пробу не вводять сторонні катіони; а самі кислоти порівняно легко видаляються з сфери реакції за умов нагрівання. Кислоти в залежності від їх природи та концентрації можуть проявляти окисні (конц. HNO3 та H2SO4) чи комплексоутворюючі властивості (HF та H3PO4). Кислоти та їх суміші, які найбільш часто використовують для розчинення представлені у таблиці 7.
Таблиця 7
Кислоти та їх суміші, які використовують під час розчинення
| Кислота | Об’єкт, що розчиняють | Примітка |
| HCl | Метали, оксиди металів, залізні руди, карбонати, органічні аміни | Кислота злетючивається під час нагрівання |
| HNO3(конц.) | Метали (крім Au, Pt, Cr, Al), сплави, сульфіди, органічні сполуки | Окисник |
| H2SO4(конц.) | Метали (сурьма, олово), оксиди металів, органічні сполуки | Окисник, можливе руйнування посуду |
| HNO3+H2SO4 | Більшість неорганічних речовин, органічні сполуки | Використовують суміш HNO3+H2SO4 змінного складу |
| HCl+HNO3 (3:1, царська горілка) | Метали (крім Au, Pt, Pd), сплави, органічні сполуки | Сильні окисні властивості |
Причиною забрудення під час розчинення можуть бути домішки у реагентах, що використовують, чи часткове розчинення матеріала посуду. Тому для розчинення застосовують кислоти високого ступеня чистоти та підбирають сосуди із відповідного матеріалу (стаканчики з платини, фторопласту, скловуглецю).
Розчинення проб часто проводять у автоклавах, що має певні переваги. Для прискорення розкладання кислотами часто використовують каталізатори. У окремих випадках для розчинення використовують ферменти.
Вибір “сухого” способу розкладання (сплавлення, зпікання, термічне розкладання) визначається задачею аналізу та природою об’єкту. “Сухий” спосіб використовують тоді, коли “мокрий” спосіб не дає задовільних результатів. Під час виконання розкладання “сухим” способом збільшується імовірність та величина похибок, особливо під час сплавлення. Це пов’язано із високою температурою обробки зразку та звідси, із більшими утратами летючих речовин та руйнуванням матеріалу посуду, а значить, із забрудненням проби. Крім того, можуть виникнути помилки внаслідок використання великого надлишку розкладаючих агентів. При цьому відбувається забрудення досліджуваного матеріалу, а розчини після обробки сплаву містять багато солей, які можуть заважати визначенню компонентів на послідуючих стадіях аналізу.
Методи розкладання окисненням і відновленням
Найпростіший метод розкладання проб з окисненням ¾ прокалювання на повітрі у відкритих чашах чи тиглях за температури 500-600°С. Такий спосіб використовують під час визначення неорганічних компонентів у органічних матеріалах, наприклад, домішок металів у біомасах та харчових продуктах. Під час визначення елементів у вигляді летучих продуктів окиснення, особливо під час елементного аналізу органічних сполук, зжигають пробу у струмі кисню чи повітря. Сухий кисень змішують при цьому з інертним газом-носієм (нітроген, гелій).
Окиснення досліджуваних речовин можна проводити у закритих скляних чи кварцевих сосудах за умов нормального та високого тиску. У цьому способі сухого озолення процес окиснення відбувається швидше та повніше.
Іноді проводять термічне розкладання проб із використанням відновлення воднем чи амоніаком. На реакції відновлення воднем у безперервному потоці суміші водню з інертним газом базується визначення кисню у металах. Цей метод розкладання використовують і в органічному аналізі під час визначення галогенів, сірки та азоту.
Таблиця 8
Термічне розкладання деяких матеріалів
| Проби | Спосіб розкладання | Температура, °С | Компонент, що визначають |
| Сульфати | Піроліз із виділенням O2 та SO2 | Сірка | |
| Фенолформальдегідні смоли | Піроліз | 300-800 | Феноли |
| Мука | Сухе озолення у відкритому сосуді | Метали | |
| Ткані тварин | Сухе озолення у відкритому сосуді з додаванням Li2CO3 | Бор | |
| Тваринні жири | Сухе озолення у відкритому сосуді з додаванням MgO | Фосфор | |
| Скло | Окиснення у тоці кисню | Сірка | |
| Органічні сполуки | Сухе озолення у тоці повітря у присутності SiO2 | 600-700 | Галогени |
| Органічні сполуки | Сухе озолення у запаяній трубці, що заповнена киснем ти містить мідь | Елементний аналіз, визначення CO2, H2O, N2. |
Аналіз сумішей катіонів і аніонів (якісний хімічний аналіз речовини)
Аналіз невідомої речовини дає можливість виявити, які катіони та аніони входять до її складу, та встановити хімічну формулу цієї речовини.
Виявлення катіонів
0,5-1 г досліджуваної речовини розчиняють у 20 мл води, якщо потрібно ¾ за умов нагрівання. Із окремих проб розчину за допомогою групових реактивів визначають, катіони яких аналітичних груп відсутні.
Дослідження на катіони першої аналітичної групи
До 0,5 мл досліджуваного розчину приливають 2 н. розчин хлоридної кислоти. Якщо утворюється осад, то в розчині були присутні катіони першої аналітичної групи.
Дослідження на катіони другої аналітичної групи.
До 0,5 мл досліджуваного розчину приливають 2 н. розчин сульфатної кислоти і нагрівають. Якщо утворюється осад, то в розчині були присутні катіони другої аналітичної групи.
Дослідження на катіони третьої аналітичної групи.
До 0,5 мл досліджуваного розчину додають надлишок натрій гідроксиду. Розчинення осаду, що утворився під час додавання перших крапель лугу, в надлишку лугу свідчить про присутність катіонів третьої аналітичної групи.
Дослідження на катіони четвертої аналітичної групи.
Якщо під час дії натрій гідроксиду на досліджуваний розчин утворюється осад, який не розчиняється у надлишку лугу, то це свідчить про наявність катіонів четвертої аналітичної групи.
Дослідження на катіони п'ятої аналітичної групи.
До 0,5 мл досліджуваного розчину додають краплями розчин амоніаку. Якщо утворюється осад, який розчиняється у надлишку амоніаку, то це свідчить про присутність катіонів п'ятої аналітичної групи
Дослідження на катіони шостої аналітичної групи.
Якщо під час проведення попередніх досліджень осад не випадав, то це свідчить про присутність катіонів шостої аналітичної групи.
Виявлення аніонів
Дослідження на аніони першої аналітичної групи.
До 0,5 мл нейтрального або слаболужного розчину добавляють 0,5 мл барій хлориду. Якщо осад випадає, то це свідчить про присутність аніонів першої аналітичної групи.
Дослідження на аніони другої аналітичної групи.
До 0,5 мл нейтрального досліджуваного розчину приливають 0,5 мл 2 н. розчину нітратної кислоти та краплями додають розчин аргентум нітрату. Випадіння осаду свідчить про наявність аніонів другої аналітичної групи.
Дослідження на аніони третьої аналітичної групи.
Якщо від дії барій хлориду та аргентум нітрату осади не випали, то, можливо, присутні аніони третьої аналітичної групи, які виявляють характерними реакціями.
Література: [9] с.550-566; [15]
МЕТОДИ РОЗПОДІЛУ І КОНЦЕНТРУВАННЯ
Лекція №16 (2 години)
План
1. Маскування.
2. Розділення і концентрування.
3. Екстракція.
4. Інші методи розділення і концентрування.
5. Хроматографія як метод розподілу і концентрування.
Маскування
Маскування ¾ гальмування чи повне уникнення хімічної реакції у присутності речовин, які здатні змінити її напрямок чи швидкість. При цьому не відбувається утворення нової фази, у чому і полягає основна перевага маскування перед розділеням, оскільки виключаються операції, що пов’язані з відділенням фаз друг від друга. Розрязняють два види маскування ¾ термічне (рівноважне) і кінетичне (нерівноважне). Під час термодинамічного маскування створюють умови, при яких умовна константа реакції знижується до такого ступеня, що реакція йде незначно. Концентрація маскуючого компонета стає недостатньою для того, щрб надійно зафіксувати аналітичний сигнал. Кінетичне маскування базується на збільшенні різниці між швидкостями маскуючої та визначаємої речовини з одним реагентом.
Можна виділити декілька груп маскуючих речовин.
1. Речовини, які утворюють з речовинами, що заважають, більш стійкі сполуки, ніж з речовинами, що визначають.
2. Речовини, що запобігають кислотно-основній реакції з утворенням малорозчинних гідроксидів.
3. Речовини, які змінюють ступінь окиснення іону, що заважає.
4. Речовини, які осаджують іони, що заважають, але осад при цьому можна не відділяти.
5. Речовини зі специфічною дією.
Іноді маскування сполучає перераховані прийоми.
Для оцінки ефективності маскування користуються індексом маскування. Це логарифм відношення загальної концентрації речовини, що заважає до її концентарції, що залишилася не зв’язаною.
У хімічному аналізі часто використовують такі маскуючі речовини:
· комплексони;
· оксикислоти;
· поліфосфати, що здатні до утворення комплексів із шестиланковою хелатною структурою (піро- і триполіфосфати натрію);
· поліаміни;
· гліцерин;
· тіосечовина;
· галогенід-, ціанід-, тіосульфат-іони;
· амоніак.
Поряд із маскуванням у хімічному аналізі іноді застосовують демаскування ¾ перевод речовини, яка замаскована у форму, що здатна вступати в реакції. Це досягається шляхом протонування маскуючої сполуки (якщо вона являється слабкою основою), незворотнім її руйнуванням чи видаленням. Демаскування можна здійснити шляхом зменшення рН розчину, окисненням маскуючої сполуки.
Розділення і концентрування
Необхідність розділення і концентрування може бути обумовлена такими факторами:
1) проба містить компоненти, які заважають визначенню;
2) концентарція компонента, що визначають, нижче межі визначення методу;
3) компоненти, які визначають, нерівномірно розподілені у пробі;
4) відсутні стандартні зразки для градуювання приладів;
5) проба високотоксична, радіоактивна чи дорога.
Розділення ¾ це операція (процес), в результаті якої компоненти, що складають вихідну суміш, відділяються один від іншого.
Концентрування ¾ операція (процес), в результаті якої збільшується відношення концентрації чи кількості мікрокомпонентів до концентрації чи кількості макрокомпонентів.
Під час розділення концентрації компонентів можуть бути близькими одна до одної, але можуть і відрізнятись. Концентрування проводять в умовах, коли концентрації компонентів різко відрязняються.
Під час концентрування речовини, які присутні у невеликій кількості, збираються у меншому об’ємі чи масі (абсолютне концентрування), чи відділяються від макрокомпонента таким чином, що відношення концентрації мікрокомпонента до концентрації макрокомпонента збільшується (відносне концентрування).
Розрізняють групове та індивідуальне виділення і концентрування: під час групового ¾ за один прийом відділяється декілька компонентів, при індивідуальному ¾ із зразка виділяють один компонент чи послідовно декілька компонентів. Під час використання багатоелементних методів визначення (атомно-емісійний, рентгенофлуоресцентний) краще використовувати групове розділення і концентрування. Під час визначення методами фотометрії, флуориметрії, атомно-абсорбційним, навпаки, краше використовувати індивідуальне виділення компонентів. Для розділення і концентрування застосовують методи екстракції, соосадження, хроматографії та інші. Хроматографію використовують головним чином для розділення складних сумішей на складові частини, соосадження ¾ під час концентрування.
Для кожної сфери хімічного аналізу є свій вибір методів розділення і концентрування. У нафтохімічній промисловості ¾ в основному хроматографічні методи, у токсікологічній хімії ¾ екстракція та хроматографія, в електронній промисловості ¾ дистиляція та екстракція.
Екстракція
Екстракція ¾ це фізико-хімічний процес розподілу речовини між двома фазами, найчастіше між двома рідинами, які не змішуються (звичайно між водою та органічним розчинником) і відповідний метод виділення, розділення та концентрування речовин.
Під час екстракції одночасно перебігають процеси: утворення сполук, які екстрагують; розподіл сполук, які екстрагують між водною та органічною фазами; реакції в органічній фазі (дисоціація, асоціація, полімеризація). Сполуку (звичайно в органічній фазі), яка відповідає за утворення сполуки, яку екстрагують, називають екстрагентом. Інертні органічні розчинники, такі як, хлороформ, бензол, що застосовують для покращення фізичних та екстракційних властивостей екстрагента, називають розріджувачами. Органічну фазу, яка відділена від водної фази і містить екстраговані сполуки, називають екстрактом. Переведення речовини з органічної фази у водну називають реекстрацією, а розчин, що використовують для реекстракції ¾ реекстрагеном.
Умови екстракції речовини
1. Для того, щоб іони металу чи інші заряджені частинки перейшли в органічну фазу, необхідно нейтралізувати заряд. Йони метала можна зв’язати у незаряджений комплекс; комплекси, що мають заряд, можна екстрагувати у вигляді іонних асоціатів.
2. Екстракція можлива, якщо розчинність сполуки, яку екстрагують, в органічному розчиннику більша, чим у воді; чим більше енергія сольватації і менше енергія гідратації, тим вище ступінь вилучання.
3. Для того, щоб сполука була гарно розчинною в органічному розчиннику, необхідно забезпечити її гідрофобність, тобто повинні, як правило, бути відсутніми групи (¾SO3H, ¾COOH, ¾OH та інші).
4. Із збільшенням розміру молекул сполуки, яку екстрагують, ступінь вилучення звичайно збільшується, оскільки великі молекули сильніше руйнують структуру води.
5. Екстракції сприяє “сольватація” молекулами екстрагента.
6. Під час екстракції іонних асоціатів мають значення заряд та розмір іонів; екстракція погіршується зі збільшенням заряду і зменшенням розміру іонів.
7. Більш стійкі комплекси екстрагуються краще.
Швидкість екстракції визначається швидкістю самого повільного процесу: чи швидкістю утворення сполук, які екстрагуються (хімічна реакція), чи швидкістю переноса різних сполук з однієї фази в іншу, чи екстракція здійснюється у змішаному режимі (швидкості обох процесів приблизно однакові).
Швидкість екстракції залежить від концентрації реагента, рН розчину, наявності маскуючих речовин, від константи дисоціації та розподілу реагента.
Класифікація екстракційних процесів
Класифікувати екстракційні процеси можна базуючих на різних ознаках:
·природа та властивості реагентів;
·тип сполуки, що переходить в органічну фазу;
·способи здійснення екстракції.
Екстракційні процеси за типом реагенту, що використовують можна розділити на три групи: екстракція кислотними (катіонообмінними), основними (аніонообмінними) та нейтральними екстрагентами (таблиця 9).
Таблиця 9
Класифікація екстракційних процесів за природою екстрагента
| Тип ектрагента | Групи сполук |
| Кислотні (катіонообмінні) | Хелатоутворюючі: диметилгліоксим, 8-оксихінолін, дифенілтіокарбазон |
| Карбонові та нафтенові кислоти | |
| Фосфорорганічні кислоти: ди(2-етилгексил)фосфорна | |
| Сульфокислоти | |
| Основні (аніонообмінні) | Солі третичних амінів |
| Солі четвертичних амонієвих основ | |
| Нейтральні (координаційні) | Ефіри: диетиловий, 2,2-дихлордиетиловий |
| Кетони: циклогесанон | |
| Фосфати | |
| Сульфіди, сульфоксиди | |
| Похідні тіосечовини |
Класифікація сполук, що екстрагують , за типом виділяє два типи сполук: неіонізовані та іонні асоціати.
За способом здійснення екстракцію підрозділяють на періодичну, безперервну, противоточну.
Інші методи розділення і концентрування
Дифузійні методи
Ці методи використовують для розділення речовин у газоподібному та рідкому станах. Молекули речовин у газоподібному та рідкому станах знаходяться у стані безперервного руху. Тому під час протікання суміші газів через пористі тела під дією градієнта тиску чи концентрації відбувається розділення частиць з різними молекулярними масами (методи трансдифузії чи дифузії відповідно).
Фільтрація
Тверді частинки, зважені у рідинах чи газах, рухаються через пористе середовище і затримуються. При цьому вдається виділяти дуже маленькі частинки з аерозолей та колоїдних розчинів.
Матеріали фільтрів різноманітні. Це папір, графіт, пористе скло, кварц, синтетичні матеріали.
Седиментація та ультрацентрифугування
Седиментиція базується на осадженні грубодисперсних частиць під дією сили тяжкості. Для осадження більш рухомих частиць невеликого розміру слід використовувати більш високе прискорення. Це здійснюється ультрацентрифугуванням.
Методи седиментації використовують для розділення речовин у колоїдному стані у вигляді суспензії.
Діаліз
Цей метод базується на різниці швидкостей проникнення різних частиць через мембрану. Якщо речовини, що розділяють ¾ іони, то можна використати різновидність методу ¾ електродіаліз (діаліз з накладанням напруги).
Хроматографія як метод розподілу і концентрування
Хроматографія - аналітичний метод, який найбільш часто використовується. Хроматографію успішно застосовують з метою дослідження у самих різних областях біології та медицини, ідентифікації антибіотиків та віднесення їх до тієї чи іншої групи антибактеріальних препаратів, для визначення найбільш важливих класів пестицидів. Такі переваги, як універсальність, експресність та чутливість роблять хроматографію важливішим аналітичним методом.
Хроматографія - це фізико-хімічний метод розподілу речовин, який базується на розподілі компонентів між двома фазами - нерухомою та рухомою. Нерухомою (стаціонарною фазою) звичайно служить тверда речовина (її часто називають сорбентом) чи плівка рідини, яка нанесена на тверду речовину. Рухома фаза - це рідина чи газ, який протікає через нерухому фазу.
Компоненти суміші, що аналізують, разом із рухомою фазою рухаються вздовж стаціонарної фази. Останню звичайно поміщують у скляну (чи металеву) трубку, яку називають колонкою. У залежності від сили взаємодії з поверхнею сорбенту (за рахунок адсорбції чи за яким-небудь іншим механізмом) компоненти переміщуються вздовж колонки з різною швидкістю. Деякі компоненти залишаються у верхньому шарі сорбенту, інші, з меншою степінню взаємодії з сорбентом, оказуються у нижній частині колонки, деякі покидають колонку разом із рухомою фазою. Таким чином компоненти розділяються.
Хроматографічні сорбційні методи розрізняють за слідуючими ознаками: за середовищем, в якому відбувається розподіл (газова, газо-рідинна, рідинна хроматографія); за механізмами розподілу (молекулярна, іонообмінна, осадочна та розподільна хроматографія); за технікою проведення розподілу (колоночна, капілярна, паперова та тонкошарова хроматографія). За метою хроматографування виділяють аналітичну хроматографію (якісний та кількісний аналіз); препаративну хроматографію (для одержання речовин у чистому вигляді, для концентрування та виділення мікродомішок); промислову хроматографію для автоматичного управління процесом (при цьому основний продукт із колонки поступає в датчик). Методами хроматографії аналізують суміші неорганічних речовин, концентрують домішки елементів. У хімічній технології хроматографію застосовують для очищення та розподілу різноманітних речовин, які є близькими за властивостями: лантаноїдів, актиноїдів, амінокислот та інших.
Тонкошарова хроматографія
Тонкошарова хроматографія проста за технікою виконання , експресна, не потребує складного обладнання. Розподіл цим методом може бути виконаний із застосуванням різних хроматографічних систем, тому виділяють адсорбційну, розподільну, іонообмінну. У методі ТСХ - різні сорбенти (алюміній(III) оксид, силікагель, целюлоза та інші), нанесені на пластинку тонким шаром. У методі застосовують хроматографічні системи рідина - твердий сорбент та рідина - рідина - твердий сорбент, крім того як рухому фазу використовують різні розчинники чи їх суміші, органічні чи неорганічні кислоти.
Компоненти, які розділяють на пластинці утворюють окремі зони (плями), розташування яких на хроматограмі характеризується величинами Rf - відносною швидкістю переміщення компонентів у тонкому шарі. Експериментально величину Rf визначають як відношення відстані x, яку пройшла речовина, до відстані L, яку пройшов розчинник від старту до лінії фронту.
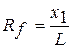 .
.
У ТСХ найчастіше використовують висхідний спосіб одержання хроматограм. Для цього застосовують скляні, металеві чи пластмасові пластинки, які вкриті тонким шаром сорбенту (нерухома фаза) звичайно товщиною 100-300 мкм. Досліджувану речовину наносять мікропіпеткою на стартову лінію та поміщають пластинку в камеру, яка містить розчинники (рухома фаза) для розділення компонентів. Хроматографування триває до тих пір, доки розчинник не пройде від лінії старту проблизно 10 см. Після чого хроматограму виймають з камери та підсушують на повітрі. Якщо утворюються безкольорові зони, то їх проявляють.
Якісний аналіз. Якщо на хроматограмі утворюються забарвлені зони, то проводять візуальні спостереження. Незабарвлені хроматограми проявляють відповідними реагентами, як правило, груповими. За характерним забарвленням кольорових зон роблять висновок про склад проби, що аналізують.
Для визначення компонентів використовують також метод, який базується на вимірюванні величин Rf для стандартної та визначаємої речовин в певному розчиннику. Ідентифікація компонентів цим способом може бути здійснена, якщо проводити хроматографування та визначення Rf для стандартного та досліджуваного розчинів в одній камері, на одній пластинці. Після проявлення хроматограм визначають Rf досліджуваного та стандартного розчинів. Порівнюють їх та роблять висновок про наявність у досліджуваному розчині тих чи інших компонентів.
Кількісний аналіз проводять безпосередньо на хроматограмі (на шарі сорбенту) за розміром плями (напівкількісне визначення).
Література: [9] с.61-69
ОБ’ЄМНИЙ (ТИТРИМЕТРИЧНИЙ) АНАЛІЗ
Лекція № 17,18 (4 години)
План
1. Об’ємний аналіз. Сутність, основні поняття, техніка та обладнання.
2. Класифікація методів об’ємного аналізу.
3. Метод нейтралізації.
4. Визначення карбонатної твердості води.
6. Редоксиметрія (окисно-відновне титрування).
7. Перманганатометрія.
8. Йодометрія.
9. Комплексонометрія.
10. Визначення загальної твердості води.
Об’ємний аналіз. Сутність, основні поняття, техніка та
обладнання
Об’ємний аналіз поєднує різноманітні методи кількісних визначень, які базуються на вимірюванні маси речовини шляхом вимірювання об’ємів розчинів взаємодіючих речовин.
Сутність титриметричного аналізу: до розчину, який приготовлено з наважки аналізуємої речовини (чи до певної частини об’єму цього розчину), поступово приливають розчин точно відомої концентрації до тих пір, доки речовини не провзаємодіють повністю. Тоді на підставі точного вимірювання об’єму реактиву розраховують вміст складової частини у досліджуваному об’єкті.
Процес приливання одного розчину до другого з метою визначення концентрації одного з них називають титруванням.
Момент закінчення реакції, коли взаємодіючі речовини повністю прореагують між собою, називають точкою еквівалентності, тому що в цей момент кількості прореагувавших речовин строго еквівалентні.
Для виконання визначень об’ємним методом необхідно мати:
·Робочий розчин (титрований чи стандартний), тобто розчин речовини, який має точно відому концентрацію.
·Посуд для точного вимірювання об’ємів розчинів речовин, що взаємодіють (бюретка, піпетка).
Криві титрування
Крива, яка виражає зміну значення рН розчину, що титрують, в залежності від об’єму прилитого робочого розчину, називають кривою титрування. На вісі абсцис відкладують об’єм прилитого робочого розчину в мл, а на вісі ординат - значення рН. Горизонтальну лінію, яка відповідає рН=7, називають лінією нейтральності, а вертикальну, що проходить через точку, яка відповідає об’єму робочого розчину, що пішов на титрування, лінією еквівалентності.
Побудувати криву титрування - це означає нанести на графік наступні точки:
· Вихідну точку титрування - точка, яка відповідає значенню рН титруємого розчину до початку титрування.
· Точку еквівалентності - точка, яка показує, при якому значенні рН титрування повинно бути закінчено.
· Нанести проміжні точки кривої.
Різка зміна рН, яка визивається додаванням останньої краплі розчину, називається стрибком титрування.
Методи встановлення точки еквівалентності
· Візуально: за зміною кольору розчину.
· Фізико-хімічними (інструментальними) методами.
Для того, щоб візуально встановити точку еквівалентності використовують індикатори.
Індикатор - речовина, яка змінює своє забарвлення у певному інтервалі значень рН середовища, в яке він введений. Більшість індикаторів - це слабкі органічні кислоти та основи (HInd чи IndOH). Якщо індикатор являється кислотою, то у розчині він дисоціює на іони HIndÛH++OH-, причому ця реакція зворотня.
Головна особливість індикаторів - їх аніони (катіони) і недисоційовані молекули мають різне забарвлення.
Згідно з іонною теорією індикаторів зміна забарвлення визивається зміщенням рівноваги дисоціації.
Згідно з хромофорною теорією індикаторів зміна їх забарвлення пов’язана із зворотнім перегрупуванням атомів в молекулі органічної сполуки. Таке зворотнє перегрупування називається таутомерією.
Зміщення рівноваги відбувається постіпово, тому забарвлення індикатору змінюється не відразу. Забарвлення стає різким коли забарвлених частиць більше дев’яноста відсотків.
Таблиця 10
Зміна забарвлення індикатора в залежності від ступеня дисоціації
| a,% | У розчині міститься | Розчин забарвлений у колір | |
| молекул | іонів | ||
| a<10 | + | - | молекул |
| 10<a<50 | + | мало | молекул з відтінком кольору іонів |
| змішаний | ||
| 50<a<90 | мало | + | іонів з відтінком кольору молекул |
| 90<a | - | + | іонів |
Інтервал переходу індикаторів
Інтервал значень рН, в яких індикатор змінює своє забарвлення називають інтервалом перехода індикатора. Розрахувати цей інтервал можна за формулою:
рН =рКінд 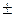 1.
1.
Класифікація методів об’ємного аналізу
В залежності від типу хімічної реакції, яка використовується для визначення, методи титриметричного аналізу класифікують таким чином:
· Метод нейтралізації.
· Окисно-відновний метод (оксидиметрія, редоксіметрія).
· Метод осадження і комплексоутворення.
Метод нейтралізації
Сутність і можливості
До методу нейтралізації відносяться усі об’ємні визначення, які базуються на реакції нейтралізації:
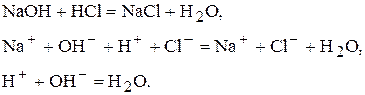
Можна проводити визначення солей, які мають сильно лужну реакцію внаслідок гідролізу. Наприклад,
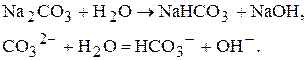
Робочі розчини
Основні робочі розчини: 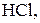
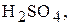
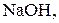
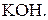 Титри цих розчинів необхідно встановлювати. Під час встановлення титрів кислот використовують буру
Титри цих розчинів необхідно встановлювати. Під час встановлення титрів кислот використовують буру 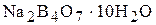 чи соду
чи соду 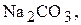 під час встановлення титрів лугів - щавлеву кислоту
під час встановлення титрів лугів - щавлеву кислоту 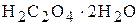 чи янтарну кислоту
чи янтарну кислоту 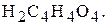
Величина рН в точці еквівалентності залежить від природи реагуючих речовин.
· Якщо кислота і основа, які вступають у взаємодію, являються сильними електролітами, то рН в точці еквівалентності дорівнює семи. Наприклад,
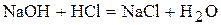 рН=7.
рН=7.
· Якщо кислота слабка , а основа сильний електроліт, то рН в точці еквівалентності буде більше семи. Наприклад,
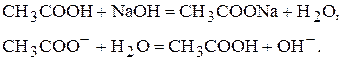 рН>7
рН>7
· Якщо кислота являється сильним електролітом, а основа - слабким, то рН в точці еквівалентності буде менше семи. Наприклад,
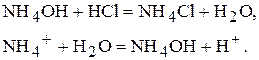 рН<7
рН<7
Індикатори в методі нейтралізації
Величина рН, до якої титрують розчин з певним індикатором, називають показником титрування цього індикатору. Найважливіші індикатори мають слідуючі області переходу та показники титрування.
Таблиця 11
Найважливіші індикатори, їх області переходу та показники титрування
| Назва індикатору | Область переходу рН | Показник титрування рТ | Забарвлення недисоційованих молекул | Забарвлення аніонів |
| Метиловий оранжевий | 3,1-4,4 | 4,0 | червоне | жовте |
| Метиловий червоний | 4,4-6,2 | 5,5 | червоне | жовте |
| Лакмус | 5,0-8,0 | 7,0 | червоне | синє |
| Фенолфталеїн | 8,0-10,0 | 9,0 | безкольорове | червоне |
Визначення карбонатної твердості води
Наявність у воді різних солей кальцію і магнію придає воді жорсткість. Солі, які можуть бути присутні - це кальцій гідрогенкарбонат, кальцій хлорид, кальцій сульфат, магній гідрогенкарбонат, магній хлорид, магній сульфат.
Види жорсткості води
· Загальна жорсткість (визначається сумою концентрацій кальцій-катіонів та магній-катіонів).
· Постійна жорсткість (обумовлена наявністю у воді таких солей: кальцій хлорид, кальцій сульфат, магній хлорид, магній сульфат).
· Тимчасова (карбонатна) жорсткість (обумовлена наявністю у воді таких солей: кальцій гідрогенкарбонат, магній гідрогенкарбонат).
· Кальцієва жорсткість (обумовлена наявністю у воді солей кальцію).
· Магнієва жорсткість (обумовлена наявністю у воді солей магнію).
Жорсткість води визначається молярною концентрацією еквівалента іонів кальцію та магнію у мілімолях на 1 дм3.
Категорії жорсткості
· М’яка (ж<4 ммоль/дм3).
· Середньої жорсткості (4<ж<8 ммоль/дм3).
· Жорстка (ж>8 ммоль/дм3).
В Україні згідно з ГОСТ 297 1-82 припустима жорсткість води для господарсько-питного водопостачання має становити не більш, ніж 7 ммоль/дм3.
Способи усунення жорсткості води
1. Методи осадження.
а) тривале кип’ятіння:
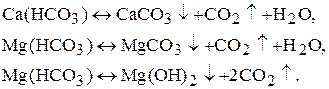
б) додавання соди або Са(OH)2:
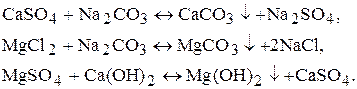
2. Метод іонного обміну.
Іоніти - речовини, які мають здатність до обміну іонів. В залежності від того, який вид іонів приймає участь в обміні їх підрозділяють на катіоніти та аніоніти.
Катіоніти здатні обмінювати катіони , а також гідроген-іони. Аніоніти здатні обмінювати аніони, а також гідроксид-іони.
Обробка води на іонітах називається обезсолюванням води. Для обезсолювання воду послідовно пропускають через катіонітовий та аніонітовий фільтри. Після використання іоніти можуть бути регенеровані.
Карбонатну жорсткість можна визначати за допомогою метода нейтралізації. Вода має слаболужну реакцію і кальцій гідрогенкарбонат та магній гідрогенкарбонат реагують з хлоридною кислотою, тому як робочий розчин під час визначення карбонатної жорсткості води беруть розчин хлоридної кислоти у присутності метилового оранжевого. При цьому перебігають реакції:
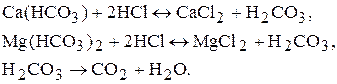
Бюретку заповнюють розчином хлоридної кислоти. У колби для титрування відміряють мірною колбою по 100 мл води, додають 3-4 краплі метилового оранжевого і титрують одержаний розчин до золотисто-рожевого забарвлення. За даними титрування розраховують карбонатну жорсткість води.
Наприклад, на титрування 100 мл води пішло 6 мл хлоридної кислоти з молярною концентрацією еквівалента 0,1 моль/л.
Дано: Результати титрування:
Сн(HCl)=0,1 моль/л V1(HCl)=5,85 мл
V(H2O)=100 мл V2(HCl)=6,05 мл
Індикатор: метиловий оранжевий V3(HCl)=6,10 мл
Жкарб=? Vсереднє(HCl)=6,00 мл
Карбонатну жорсткість можна розрахувати за рівнянням:
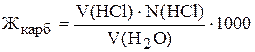 ,
,
де V(HCl) ¾ середнє значення об’єму хлоридної кислоти, яке пішло на титрування, мл;
N(HCl) ¾ молярна концентрація еквівалента розчину хлоридної кислоти, моль/л;
V(H2O) ¾ об’єм води, мл.
Підставляємо одержані дані титрування:
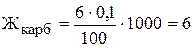 (мг-екв)/л.
(мг-екв)/л.
Редоксиметрія (окисно-відновне титрування)
Загальна характеристика методів окиснення-відновлення
Ці методи поєднюють різні об’ємно-аналітичні визначення, які пов’язані з застосуванням реакцій окиснення та відновлення.
За допомогою титрованих розчинів окисників визначають кількісний вміст відновників (наприклад, ферум(ІІ)-катіони, манган(ІІ)-катіони). За допомогою титрованих розчинів відновників визначають окисники (наприклад, ферум(ІІІ)-катіони, купрум(ІІ)-катіони, хлор, бром і т.д.).
Як індикатори в методах окиснення-відновлення застосовують різні органічні сполуки, які самі являються окисниками чи відновниками. Крім того, для деяких реакцій оксидиметрії підібрані специфічні реактиви, які змінюють забарвлення в еквівалентній точці даного титрування. Наприклад, в йодометрії таким специфічним і чутливим реактивом є крохмаль, який стає синім у присутності йода.
В усіх випадках окисно-відновних реакцій окисники та відновники реагують між собою у кількостях, які відповідають їх еквівалентам. Тому, для находження грам-еквівалента окисника необхідно грамм-молекулу окисника поділити на число електронів, одержаних під час реакції однією молекулою, а для визначення грамм-еквівалента відновника слід грамм-молекулу його поділити на число електронів, які були віддані під час реакції однією молекулою відновника:
Е= 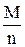 ,
,
де Е - грам-еквівалент окисника чи відновника;
n - число електронів, які були приєднані однією молекулою окисника, чи число електронів, які були віддані однією молекулою відновника;
М - грам-молекула окисника чи відновника.
Реакції окиснення-відновлення перебігають складніше, чим реакції, які застосовують в методі нейтралізації. Вкажемо основні особливості окисно-відновних реакцій:
а) у багатьох реакціях взаємодіють не тільки окисники та відновники, але й речовини, які не змінюють своєї валентності, наприклад, кислоти, луги;
б) реакції перебігають у декілька стадій, причому кожна з цих стадій перебігає з різною швидкістю;
в) у процесі реакції часто утворюються речовини, які змінюють хід самої реакції;
г) швидкість реакції нижче швидкості обмінних реакцій;
д) багато реакцій є зворотніми.
Реакції окиснення-відновлення, на основі яких здійснюється кількісний аналіз, повинні відповідати наступним вимогам:
· Окисно-відновна реакція повинна перебігати в необхідному напрямку і бути практично незворотньою. Напрямок окисно-відновної реакції залежить від величини окисно-відновних потенціалів реагуючих речовин.
· Окисно-відновні реакції повинні перебігати з достатньою швидкістю. Швидкість реакції можна збільшити збільшенням температури, концентрації реагуючих речовин, зміною рН розчину і застосуванням каталізаторів.
Переваги окисно-відновних методів
· Велика точність, простота, гарна відтворюваність.
· Експрессність.
· Можливість визначати не тільки окисники та відновники, але й сполуки, які не підлягають окисненню і відновленню.
· Велика різноманітність.
· Титрування можна здійснювати індикаторним, безіндикаторним та інструментальним методами.
· Визначення можна проводить методами прямого, зворотнього та непрямого титрування.
· Деякі окисники та відновники є хімічно чистими, тобто можна приготувати стандартний розчин за точною наважкою
· Можна титрувати в усіх середовищах.
Класифікація окисно-відновних методів титрування
У залежності від характеру титранта окисно-відновні методи титрування класифікують таким чином:
1. Перманганатометрія.
2. Іодометрія.
3. Аскорбінометрія.
4. Титанометрія і т. д.
5.
Перманганатометрія
Сутність метода перманганатометрії
Перманганатометрія ¾ метод об’ємного аналізу, робочим розчином якого являється розчин калій перманганату. У процесі титрування досліджуваного розчину малиново-фіолетове забарвлення розчину калій перманганату знебарвлюється. Однак, після досягнення точки еквівалентності перша надлишкова крапля розчину калій перманганату забарвлює рідину, що титрують, у блідо-малиновий колір. Індикатором у даному випадку являється сам калій перманганат. Тому під час перманганатометричних визначень сторонні індикатори не додають.
Калій перманаганат проявляє окисні властивості у кислому та лужному (чи нейтральному) середовищі. Під час титрування кислих розчинів манган(VII)-катіон, який входить до складу калій перманганату, відновлюється до безбарвних манган(ІІ)-катіонів:
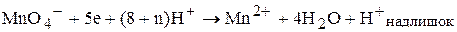 .
.
Під час титрування у нейтральному або лужному середовищі манган(VII)-катіон відновлюється тільки до манган(IV)-катіонів, наприклад до манган діоксиду:
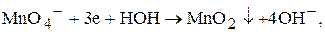
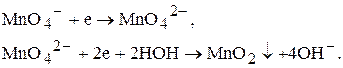
Речовини, які можна визначати методом перманганатометрії
· Відновники: метали (Fe, Bi, Ag, Cd, Zn та ін.),
неметали (Sb, As, P та ін.),
йони (Fe2+, Cr2+, Mn2+, Cl-, Br-, I-, S2- та ін.).
· Окисники: Fe3+, Cr(VI), NO3-, ClO3-, BrO3-, IO3-, PbO2, MnO2 та ін.
Переваги методу
1. Титрування проводиться без індикатору.
2. Можна титрувати у кислому та лужному середовищах.
3. Високий потенціал Е0(MnO4-/Mn2+)=1,52 В.
4. Калій перманганат ¾ дешевий та доступний реагент.
Недоліки методу
1. Розчин калій перманганату є встановленим.
2. Неможна проводити титрування у присутності хлорид-іонів.
3. Деякі реакції перебігають повільно за кімнатною температурою, тому необхідно застосовувати нагрівання.
4. Необхідно сторого дотримуватись умов, які рекомендує методика аналізу.
Йодометрія
Сутність методу йодометрії
Атоми йоду, як і інших галогенів, мають властивість віднімати електрони у речовин-відновників. Тому елементарний йод звичайно веде себе в реакціях як окисник: I2+2e=2I-. Аніони I- , навпаки, легко віддають свої електрони речовинам-окисникам і відіграють у реакціях роль відновників: 2I--2е=I2. Ці властивості йоду та його йодид-іонів лежать в основі йодометрії. Відомо, що вільний йод забарвлює крохмаль у синій колір. Якщо до розчину якого-небудь відновника додати крохмаль та титрувати йодом, то після досягнення точки еквівалентності надлишкова краплина йоду викличе синє забарвлення, що не зникає. Можна робити і навпаки, тобто до розчину йода у присутності крохмалю поступово приливати відновник. У цьому випадку точку еквівалентності визначають за знебарвленням синього кольору.
Йод малорозчинний у воді, тому, як стандартний розчин, використовують його розчин у калій йодиді: I2+I-Û[І3]-.
Еквівалент йоду розраховують за формулою: Е(I2)= 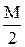 .
.
Е(І2/2І-)=0,53 В. Це означає, що йод окиснює менше число відновників, ніж калій перманганат.
Методи йодометричного титрування
1. Метод прямого титрування
Речовини, що легко окиснюються йодом, титрують розчином йоду в калій йодиді К[І3]. Таким чином визначають сульфіди, тіосульфати, сульфати та ін.
2. Метод зворотнього титрування
Речовини, які важче окиснюються йодом, обробляють розчином К[І3] і потім через деякий час відтитровують надлишок К[І3] стандартним розчином Na2S2O3.
3. Метод непрямого титрування
Речовини, які відносяться до окисників, обробляють розчином калій йодиду, а потім йод, що виділився, відтитровують розчином Na2S2O3. Цим методом визначають перманганати, хромати, біхромати, йодати та ін.
Визначення відновників
Під час визначення відновників як робочий розчин застосовують розчин йоду. Прикладом такої реакції може бути взаємодія між натрій тіосульфатом та йодом:
2Na2S2O3+I2®2NaI+Na2S4O6
I2+2e®2I- 1 відновлення, окисник
2S2O32- -2e®S4O62- 1 окиснення, відновник
I2+2S2O32-®2I-+S4O62-
Е(Na2S2O3×5Н2О)=М=248,2 г/моль; Е(I2)=М/2=126,8 г/моль.
Концентрацію натрій тіосульфату розраховують за формулою:
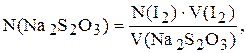
де N(І2) ¾ молярна концентрація еквіваленту розчину йоду, моль/л;
V(І2) ¾ об’єм розчину йоду, який пішов на титрування, мл;
V(Na2S2O3) ¾ об’єм розчину, який взяли для титрування, мл.
Визначення окисників
Під час визначення окисників не можна титрувати розчином калій йодиду внаслідок неможливості фіксування точки еквівалентності. Під час титруваннґ будь-якого окисника, наприклад K2Cr2O7, кінець реакції K2Cr2O7+6KI+14HCl=3I2+8KCl+
+2CrCl3+7H2O характеризовався би припиненням утворення вільного йоду. Але цей момент помітити не можна. Тому в цьому випадку застосовують непрямий метод ¾ метод заміщення. До суміші розчину калій йодиду та кислоти додають точно відомий об’єм розчину досліджуваного окисника, наприклад K2Cr2O7. Для завершення реакції розчин залишають на 5 хвилин, після чого йод, який виділився, відтитровують натрій тіосульфатом. Еквівалентну точку фіксують за допомогою розчину крохмалю, який з йодом утворює синє забарвлення.
Концентрацію калій дихромату розраховують за формулою:
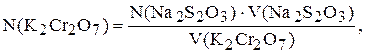
де N(Na2S2O3) ¾ молярна концентрація еквіваленту розчину натрій тіосульфату, моль/л;
V(Na2S2O3) ¾ об’єм розчину, який пішов на титрування, мл;
V(K2Cr2O7) ¾ об’єм розчину, який взяли для титрування, мл.
Переваги методу
1. Висока точність.
2. Розчини йода забарвлені, тому можна здійснювати титрування без індикаторів.
3. Моживість застосування неводних розчинів йоду.
4. Можливість визначення сполук, які безпосередньо з йодом та йодидами не реагують.
Недоліки методу
1. Можливість втрати йоду внаслідок його леткості (слід проводити реакцію у колбах з притертим корком).
2. Можливість окиснення йодид-іонів, що сприяє зменшенню рН розчину та дія сонячного світла, тому титрування слід проводити за помірної кислотності розчину та розчини йода необхідно зберігати у темних склянках чи у зачинених шафах.
3. Неможна проводити титрування у лужному середовищі (гідроксид-іони визивають реакцію диспропорціонування йоду).
4. Відносно повільна швидкість реакції.
5. Можливість адсорбції йоду.
6. Необхідність точно дотримуватись методик визначення.
Комплексонометрія
Комплексонометричне титрування базується на реакціях утворення комплексів іонів металів з амінополікарбоновими кислотами (комплексонами). Із великої кількості амінополікарбонових кислот найбільш часто використовують етилендіамінтетраетанову кислоту (Н4Y)
НООС¾Н2С СН2¾СООН
½ ½
NH+¾CH2¾CH2¾NH+
½ ½
-ООС¾Н2С СН2¾СОО-.
Внаслідок низької розчинності у воді сама кислота не підходить для приготування розчину титранта. Для цього звичайно використовують дигідрат її двунатрієвої солі Na2H2Y×2H2O (ЕДТА). Цю сіль можна одержати додаванням до суспензії кислоти натрій гідроксиду до рН»5. У більшості випадків для приготування розчину ЕДТА використовують комерційний препарат, а потім розчин стандартизують. Можна також використовувати фіксанал ЕДТА.
Реакції взаємодії різних катіонів з ЕДТА у розчині можна представити рівняннями:
Са2++ H2Y2-=СаY2-+2Н+,
Ві3++ H2Y2-=ВіY-+2Н+,
Th4++ H2Y2-=ThY+2Н+.
Видно, що незалежно від заряду катіона утворюються комплекси з співвідношенням компонентів 1:1. Значить, молекулярна маса еквівалента ЕДТА і досліджуваного іона метала дорівнюють їх молекулярним масам.
Ступінь перебігу реакції залежить від рН та константи стійкості комплексонату. Катіони, що утворюють стійкі комплексонати, наприклад Fe(III), можуть бути відтитровані у кислих розчинах. Йони Са(ІІ), Mg(II) та інші, що утворюють менш стійкі комплексонати, титрують за умови рН=9 і вище.
Фіксування точки еквівалентності
5. Реакції комплексоутворення супроводжуються виділенням гідроген-іонів у кількості, еквівалентній катіонам, тому для фіксування точки еквівалентності можна використовувати кислотно-основні індикатори.
6. Застосування фізико-хімічних методів.
7. Використання специфічних індикаторів-комплексоутворювачей, які утворюють з катіонами забарвлені комплексні сполуки (метал-індикатори).
а) еріохром чорний Т (С20H13O7N3S);
б) мурексид (С8Н6N6O6).
Дата добавления: 2016-12-16; просмотров: 1796;