Агрегатний стан речовин
У залежності від умов існування речовини можуть знаходитися у твердому, рідкому, газоподібному стані або у вигляді плазми. У газоподібному стані речовини заповнюють увесь об'єм посудини, в якій знаходяться. Вони мають здатність стискатися і утворюють однорідні суміші. Молекули у газах рухаються хаотично. Для ідеальних газів, в яких не враховується міжмолекулярна взаємодія, тиск р, об'єм V і число молів газу n зв'язані між собою рівнянням Мендєлєєва-Клапейрона:
pV = nRT. , (3.1)
Під час охолодження газ конденсується перетворюючись на рідину. Відстань між молекулами в рідинах значно менша, ніж у газах. Молекули вільно переміщуються відносно одна одної, тому рідини здатні текти і приймати форму посудини, в якій вони поміщені. Тепловий рух молекул призводить до їх неупорядкованого розташування. У той же час сусідні часточки розташовуються у рідині не хаотично, а більш-менш упорядковано. Рідини характеризуються ізотропністю, тобто ідентичністю властивістю у будь-якій точці і тотожністю у будь-якому напрямку.
У твердому агрегатному стані середня відстань між часточками дорівнює їх розмірам, а енергія їх взаємодії значно більша, ніж у рідинах. Основним видом руху часточок є коливальний рух поблизу фіксованого рівноважного положення. Тверді речовини можуть бути в аморфному або кристалічному стані. В аморфному стані структура речовини не упорядкована. Аморфна речовина ізотропна, тобто її можна розглядати як переохолоджену рідину. Плавляться такі речовини не за певної температури, а у деякому інтервалі температур. Аморфними речовинами є скло, органічні полімери. Більшість твердих тіл є кристалами – системами, в яких часточки, що їх утворюють (атоми або іони) розташовані у тримірному просторі регулярно і упорядковано. На відміну від аморфних речовин у кристалах значення параметрів будуть різними у різних напрямках, тобто кристали анізотропні. Перехід кристалів із твердого стану в рідкий відбувається стрибкоподібно за певної температури, яка називається температурою плавлення.
Типовими представниками кристалічних речовин є метали, деякі мінерали. На лекції надається характеристика структури і загальних фізико-хімічних властивостей металів: наявності металічного блиску, пластичності, здатності проводити тепло і струм.
3.2Фазові рівноваги
Термодинамічні системи поділяються на гомогенні і гетерогенні. Система, що складається лише з однієї фази називається гомогенною, якщо ж у системі є дві і більше фаз – гетерогенною. Фазою називають частину системи, яка має однакові фізико-хімічні властивості і відокремлена від інших фаз видимою межею поділу. Компонентом називається хімічно індивідуальна речовина, яка будучі виділена із системи може існувати у вільному стані тривалий час. Фазовими переходами називаються процеси переходу компонентів з однієї фази в іншу, які не супроводжуються хімічними реакціями, наприклад процеси плавлення, конденсації, розчинення. За незмінності зовнішніх умов у системі встановлюється фазова рівновага – стан коли швидкість переходу компонентів із однієї фази в іншу дорівнює швидкості їх зворотного переходу.
Основним законом фазової рівноваги є правило фаз Гіббса:
С = К – Ф + n, (3.2)
де Ф – число фаз у системі; К – число незалежних компонентів; n – число зовнішніх факторів, що впливають на рівновагу в системі; С – число ступенів вільності, яке дорівнює числу факторів, які можна довільно змінювати у певних межах, не змінюючи при цьому фазовий стан системи. Число незалежних компонентів – це мінімальна кількість компонентів, яка необхідна для утворення усіх фаз системи.
Можливість і напрямок фазових переходів речовин визначається величиною їх хімічних потенціалів m – функції, що характеризує стан компонента у фазі за певних зовнішніх умов. Вільна енергія відкритих систем залежить не тільки від їх температури, тиску і об'єму, але й від кількості речовини компонентів у системі. При доданні у систему або вилучені з неї певної кількості речовини будь-якого компоненту її внутрішня енергія, а відповідно, і вільна енергія будуть змінюватися.
Хімічний потенціал компоненту визначається за зміною значень термодинамічних потенціалів системи під час додання до неї (або вилучення) 1 моля цього компоненту. Математично це визначення можна записати:
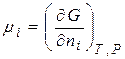 =
= 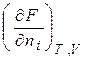 . (3.3)
. (3.3)
В інтегральному вигляді рівняння (4.3.) буде мати вигляд:
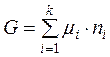 . (3.4)
. (3.4)
Тобто термодинамічні потенціали систем дорівнюють сумі добутків хімічних потенціалів усіх компонентів системи на їх кількість за умов сталості певних параметрів: температури, об’єму, тиску.
Хімічний потенціал залежить від концентрації компоненту у системі і за сталої температури визначається рівнянням:
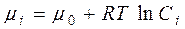 , (3.5)
, (3.5)
де Сі – концентрація і-го компоненту у розчині, m0 – стандартний хімічний потенціал компоненту (значення хімічного потенціалу за стандартних умов).
Самочинний перехід компоненту здійснюється з фази, де його хімічний потенціал більший, у фазу, де його потенціал менший. Перехід супроводжується зменшенням значення хімічного потенціалу компоненту у першій фазі і збільшенням його у другій. Внаслідок чого різниця між хімічними потенціалами компонентів в обох фазах зменшується, і коли значення хімічних потенціалів зрівняються, досягається стан фазової рівноваги. Таким чином, хімічний потенціал є критерієм, що характеризує здатність компонентів до виходу з фази шляхом випаровування, кристалізації, розчинення тощо.
3.3 Фізико-хімічний аналіз
Задачею фізико-хімічного аналізу (ФХА) є вивчення залежностей між фізичними властивостями рівноважних систем та їх складом і зовнішніми факторами, що впливають на стан рівноваги. Основним принципом ФХА є принцип безперервності М.С.Курнакова: "При поступовій неперервній зміні параметрів системи (тиску, температури або складу фаз) властивості системи також змінюється безперервно; якщо ж одна з фаз зникає або з’являється нова, то властивості системи змінюються стрибкоподібно". На підставі дослідницьких результатів будують діаграми стану (фазові діаграми), які графічно описують одержані залежності. На цих діаграмах широко застосовуються геометричні поняття: окремі фази являють собою частину простору, двофазні рівноважні системи – лінії і, трифазні системи зображують крапками. Аналіз фазових діаграм дозволяє виявляти наявність у системі хімічних сполук, їх склад, стійкість, температури фазових переходів тощо.
 Діаграма стану води. Розглянемо діаграму стану однокомпонентної системи на прикладі діаграми стану води (див. рис. 3.1), яка характеризує зміну властивостей води у залежності від температури і тиску. Криві на діаграмі поділяють площину між координатними осями на 3 області: нижче від лінії МОК – область існування ненасиченої пари, між лініями ОL і ОK – рідкої води, ліворуч від лінії МОL – льоду. У межах кожної з цих областей можна довільно змінювати тиск і температуру – система при цьому залишиться однофазною. Система за наявності в ній одночасно двох фаз моноваріантна, тобто в ній довільно можна змінювати лише один з параметрів, інший повинен змінюватись у залежності від першого. Лінії ОК і МО – це криві випаровування і возгонки, вздовж яких з підвищенням температури або тиску здійснюються процеси випаровування і возгонки відповідно. У разі зниження температури або тиску вздовж цих кривих відбуваються оборотні процеси конденсації пари, тобто перехід її в рідину або тверду фазу. Лінія ОL – характеризує процеси плавлення льоду та кристалізації рідкої води. Точка О називається нульовою точкою води: за умов, що відповідають цій точці, у рівновазі одночасно знаходяться три фази – рідка вода, лід і водяна пара. Система за цих умов – безваріантна.
Діаграма стану води. Розглянемо діаграму стану однокомпонентної системи на прикладі діаграми стану води (див. рис. 3.1), яка характеризує зміну властивостей води у залежності від температури і тиску. Криві на діаграмі поділяють площину між координатними осями на 3 області: нижче від лінії МОК – область існування ненасиченої пари, між лініями ОL і ОK – рідкої води, ліворуч від лінії МОL – льоду. У межах кожної з цих областей можна довільно змінювати тиск і температуру – система при цьому залишиться однофазною. Система за наявності в ній одночасно двох фаз моноваріантна, тобто в ній довільно можна змінювати лише один з параметрів, інший повинен змінюватись у залежності від першого. Лінії ОК і МО – це криві випаровування і возгонки, вздовж яких з підвищенням температури або тиску здійснюються процеси випаровування і возгонки відповідно. У разі зниження температури або тиску вздовж цих кривих відбуваються оборотні процеси конденсації пари, тобто перехід її в рідину або тверду фазу. Лінія ОL – характеризує процеси плавлення льоду та кристалізації рідкої води. Точка О називається нульовою точкою води: за умов, що відповідають цій точці, у рівновазі одночасно знаходяться три фази – рідка вода, лід і водяна пара. Система за цих умов – безваріантна.
Рисунок 3.1 – Діаграма стану води
Лекція №4
3агальна характеристика розчинів
План лекції
1. Розчинність речовин. Механізм процесу розчинення.
2. Дифузія. Закон Фіка.
3. Властивості розчинників. Полярні і неполярні розчинники.
4. Способи вираження концентрації розчинів.
Рекомендована література: [1] С.290-323, [2] С.49-51, [3] С.76-83, [4] С.151-155.
4.1 Розчинність речовин
Розчини – це гомогенні системи, в яких речовини розподілені в середовищі іншої речовини, яку називають розчинником, у вигляді молекул або іонів. Під розчиненням розуміють процес взаємодії речовини з рідиною, який не супроводжується їх хімічною взаємодією і дає можливість після видалення розчинника одержати вихідну речовину у незмінному стані. Розчинність речовин виражають максимальним числом грамів речовини, які можна розчинити у 100 г розчинника. Процес розчинення являє собою низку таких послідовних процесів:
· руйнування структури розчиненої речовини (наприклад кристалічних ґраток твердих речовин) і утворення іонів, молекул або атомів;
· взаємодія молекул розчинника з частинками розчиненої речовини – процес сольватації (у випадку коли розчинник вода – процес називається гідратацією);
· розподіл сольватованих часточок в об’ємі розчинника – процес дифузії.
4.2 Дифузія
Дифузія – це самочинний процес вирівнювання концентрації речовин у розчині внаслідок безладного теплового руху часточок. Процес дифузії необоротний: речовина переноситься з частини розчину, де її концентрація більша, у ту частину, де вона менша і припиняється після вирівнювання концентрації.
Швидкість дифузії визначається за законом Фіка: “Маса речовини dm, що переноситься шляхом дифузії пропорційна площині S, через яку проходить дифузія, часу dτ та градієнту (різниці) концентрацій dC/dx”:
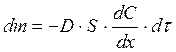 . (4.1)
. (4.1)
Здатність речовин до дифузії характеризується їх коефіцієнтом дифузії – D.
4.3 Властивості розчинників
Під час вибору ефективних розчинників користуються таким правилом: у неполярному розчиннику добре розчиняються неполярні речовини, погано розчиняються речовини з помітною полярністю і зовсім не розчиняються речовини іонного типу. Для полярних розчинників спостерігається протилежна залежність.
Полярними розчинниками, в яких розчиняються білки, вуглеводи, мінеральні речовини з іонними або ковалентними полярними зв'язками, є вода, аліфатичні спирти, естери, ацетон, рідкі органічні кислоти тощо. До неполярних розчинників, в яких розчиняються жири, жиророзчинні вітаміни, відносять рідкі аліфатичні вуглеводні та їх галогенопохідні, бензол тощо.
На лекції наводяться приклади розчинності речовин у залежності від їх природи, надається характеристика найбільш поширених розчинників.
Процес розчинення перебігає самочинно і завершується утворенням насиченого розчину, в якому міститься максимально можлива за даної температури кількість розчиненої речовини. Насичений розчин знаходиться в рівновазі з осадом розчиненої речовини. У ненасиченому розчині міститься менше розчиненої речовини, ніж у насиченому, а в пересиченому – більше. Останній є нестійкою системою, з якої розчинена речовина випадає у вигляді кристалів. При цьому із водних розчинів часто випадають кристали, що містять у собі молекули води. Кристалічні речовини, до складу яких входить певне число молекул води, називають кристалогідратами. Значна частина природних мінералів існує у вигляді кристалогідратів: CаSO4×2H2O – гіпс, Na2SO4×10H2O – глауберова сіль.
4.4 Способи вираження концентрації розчинів.
Важливою характеристикою розчинів є концентрація в них розчинених речовин. Відомі такі способи вираження концентрації.
Молярна концентрація, С (моль/л) – кількість молів розчиненої речовини, що міститься в одиниці об’єму розчину:
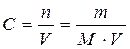 , (4.2)
, (4.2)
де n – кількість розчиненої речовини, моль; m – маса розчиненої речовини, г; V – об'єм розчину, л; М – молярна маса розчиненої речовини, г/моль.
Моляльна концентрація, b (моль/кг) – кількість молів розчиненої речовини, що міститься в одиниці маси розчинника:
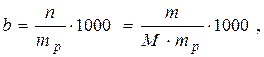 (4.3)
(4.3)
де n – кількість розчиненої речовини, моль; m – маса розчиненої речовини, г; mр – маса розчину, г; М – молярна маса розчиненої речовини, г/моль.
Мольна частка розчиненої речовини, Х – відношення кількості речовини до сумарної кількості молів усіх компонентів розчину, включаючи розчинник:
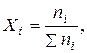 (4.4)
(4.4)
де nі – кількість молів розчиненої речовини, концентрацію якої визначають.
Масова частка, w – відношення маси розчиненої речовини до маси розчину.
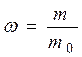 , (4.5)
, (4.5)
де m – маса розчиненої речовини В, г; m0 – маса розчину, г.
Якщо відомі об’єм розчину V в мл і його густина r в г/мл, то масову частку розчиненої речовини можна розрахувати користуючись рівнянням:
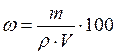 %. (4.6)
%. (4.6)
На лекції надаються приклади розрахунків концентрації розчинів.
Лекція №5
Фізико-хімічні властивості розчинів
План лекції
1. Розчини газів у рідинах. Закон Генрі.
2. Закон Рауля. Відхилення від закону Рауля для неідеальних систем.
3. Ебуліоскопічний та кріоскопічний методи аналізу.
4. Осмос. Осмотичний тиск. Закон Вант-Гоффа.
Рекомендована література: [1] С.295-312, [2] С.51-54, [3] С.82-91, [4] С.155-160.
5.1 Розчини газів у рідинах
За своїми властивостями і природою розчини газів у рідинах аналогічні усім іншим рідким розчинам. Якщо хімічна взаємодія між газом і рідиною відсутня, то концентрація газу в рідині незначна. Для таких систем розчинність газів підкоряється закону Генрі: "За сталої температури концентрація газу в рідині прямо пропорційна тиску цього газу над розчином".
C = k×P, (5.1)
де C – концентрація газу в рідині, моль/л; P – тиск газу над розчином; k – коефіцієнт, що залежить від природи газу і рідини.
Розчинність газів виражають в об’ємі газу, що розчиняється в одиниці об’єму рідини – мл/л. Розчинення газів є екзотермічним процесом, тому з підвищенням температури розчинність газів зменшується. На розчинність газів впливає також присутність у рідині домішок: у розчинах розчинність газів менша ніж у чистій воді.
Закон Рауля
Важливою характеристикою рідини є тиск її насиченої пари. Для чистої рідини тиск насиченої пари залежить тільки від температури, для її розчинів – від температури і концентрації. Рауль встановив, що тиск насиченої пари розчинника над розчином завжди менший, ніж над чистим розчинником, і його відносне зниження дорівнює мольній частці розчиненої речовини.Тобто:
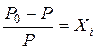 , (5.2)
, (5.2)
де Р0 – тиск насиченої пари розчинника на чистим розчинником, Р – тиск насиченої пари розчинника над розчином, Хі – мольна частка розчиненої речовини.
Загальний тиск насиченої пари над розчином двох летких речовин дорівнює сумі парціальних тисків обох компонентів, тобто:
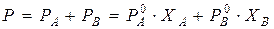 , (7.3)
, (7.3)
де РА і РВ – парціальні тиски насиченої пари компонентів А і В над їх розчином; 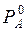 і
і 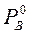 – тиски насиченої пари чистих компонентів; ХА і ХВ – мольні частки компонентів А і В у розчині. Залежність парціальних тисків насиченої пари компонентів і загального тиску над розчином зображена на рис. 5.1. В ідеальних розчинах тиск насиченої пари компонентів над розчином є лінійною функцією від складу розчину: зміна тиску пари у таких системах показана пунктирними лініями. Реальні розчини не підкоряються закону Рауля. Відхилення бувають позитивні – у бік більших значень і негативні – у бік менших значень.
– тиски насиченої пари чистих компонентів; ХА і ХВ – мольні частки компонентів А і В у розчині. Залежність парціальних тисків насиченої пари компонентів і загального тиску над розчином зображена на рис. 5.1. В ідеальних розчинах тиск насиченої пари компонентів над розчином є лінійною функцією від складу розчину: зміна тиску пари у таких системах показана пунктирними лініями. Реальні розчини не підкоряються закону Рауля. Відхилення бувають позитивні – у бік більших значень і негативні – у бік менших значень.
Так, у системі етанол-  гексан мають місце позитивні відхилення. Під час змішування цих речовин асоціати спирту розпадаються, тобто процес супроводжується поглинанням теплоти. Негативні відхилення мають місце у таких системах, як етанол-вода, де під час розчинення утворюються асоціати молекул, і спостерігається виділення теплоти.
гексан мають місце позитивні відхилення. Під час змішування цих речовин асоціати спирту розпадаються, тобто процес супроводжується поглинанням теплоти. Негативні відхилення мають місце у таких системах, як етанол-вода, де під час розчинення утворюються асоціати молекул, і спостерігається виділення теплоти.
Рисунок 5.1 – Залежність тисків насиченої пари летких компонентів від концентрації розчину: а – позитивні відхилення від закону Рауля; б – негативні відхилення
5.3 Ебуліоскопічний та кріоскопічний методи аналізу
Рідина закипає за температур, за яких тиск її насиченої пари дорівнює зовнішньому тиску. Із закону Рауля виходить, що для досягнення температури кипіння розчину його необхідно нагріти до більш високої температури, ніж чистий розчинник, при цьому підвищення температури кипіння буде прямо пропорційно моляльній концентрації розчину:
Ткип– Т0кип= DТкип= Е×b,(5.4)
де Ткип і Т0кип – температури кипіння розчину і розчинника, відповідно; b –моляльна концентрація розчину, Е – ебуліоскопічна стала розчинника.
Замерзають розчини за більш низьких температур, ніж чисті розчинники:
Т0зам– Тзам= DТзам= Ккр×b,(5.5)
де Т0зам і Тзам – температури замерзання розчинника і розчину, відповідно; b –моляльна концентрація розчину, Ккр – кріоскопічна стала розчинника.
Методи дослідження речовин, що ґрунтуються на визначенні температур кипіння і кристалізації їх розчинів називають ебуліоскопією і кріоскопією.
Осмос
Якщо розчинник і розчин або два розчини різної концентрації розділити напівпроникною мембраною, крізь яку проходять молекули розчинника і затримуються часточки розчиненої речовини, то спостерігається дифузія розчинника в розчин через мембрану. Такий однобічний перехід розчинника у більш концентрований розчин називається осмосом. При цьому в розчині виникає осмотичний тиск, який визначають за законом Вант-Гоффа:
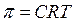 , (5.6)
, (5.6)
де С – молярна концентрація розчину.
У розчинах спостерігається зморщування клітин – плазмоліз, зумовлене втратою води, яка виходить з клітин у більш концентрований зовнішній розчин. Якщо ж помістити клітину у чисту воду, то остання всмоктується у клітину. Об’єм клітини збільшується і вона перебуває у стані напруження – тургору. Розчини цукру або солі застосовують для зберігання харчових продуктів: бактерії, що викликають їх псування, зазнають у розчинах плазмолізу і гинуть.
Лекція № 6
Методи розділення рідких систем
План лекції
1. Взаємна розчинність рідин. Обмежена взаємна розчинністю рідин.
2. Екстракція. Закон Нернста-Шилова.
3. Фракційна перегонка рідких сумішей летких речовин. Закони Коновалова.
Рекомендована література: [1] С.310-318, 324-328, [2] С.54-55, [3] С.80-81.
6.1 Взаємна розчинність рідин
 Під час змішування двох рідин з обмеженою взаємною розчинністю система розшаровується на два насичених розчини. За сталої температури вони мають строго визначений склад, який не змінюється після додання у систему нових порцій рідин, змінюється лише відносні кількості розчинів. На рис. 6.1 наведено діаграму, що характеризує взаємну розчинність води і бутанолу. Криву аКb називають кривою розшаровування. Вона розділяє області існування гомогенної та гетерогенних систем. Точка К відповідає верхній критичній температурі розчинення. Будь-які точки, що лежать поза кривою аКb, відповідають рідкому розчину. Якщо ж фігуративна точка лежить усередині області розшаровування,то система розшаровується на два розчини, склад яких визначається точками перетину нод-ізотерм з кривою розшаровування.
Під час змішування двох рідин з обмеженою взаємною розчинністю система розшаровується на два насичених розчини. За сталої температури вони мають строго визначений склад, який не змінюється після додання у систему нових порцій рідин, змінюється лише відносні кількості розчинів. На рис. 6.1 наведено діаграму, що характеризує взаємну розчинність води і бутанолу. Криву аКb називають кривою розшаровування. Вона розділяє області існування гомогенної та гетерогенних систем. Точка К відповідає верхній критичній температурі розчинення. Будь-які точки, що лежать поза кривою аКb, відповідають рідкому розчину. Якщо ж фігуративна точка лежить усередині області розшаровування,то система розшаровується на два розчини, склад яких визначається точками перетину нод-ізотерм з кривою розшаровування.
Рисунок 6.1– Діаграма стану системи вода-бутанол.
6.2 Екстракція
Якщо до системи, що складається з двох взаємно нерозчинних рідин, додати речовину, яка розчиняється в обох рідинах, то утворюється система двох розчинів цієї речовини з концентраціями С1 і С2. Для такої системи визначено, що відношення концентрацій розчиненої речовини у двох рідких фазах за сталої температури є величиною сталою незалежно від загальної кількості цієї речовини. Цю залежність називають законом Нернста-Шилова:
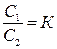 , (6.1)
, (6.1)
де К – коефіцієнт розподілу. Закон розподілу дає змогу обґрунтувати процес екстракції – вилучення розчиненої речовини з розчину шляхом додання до нього іншого розчинника. Екстракцію звичайно здійснюють у декілька циклів:
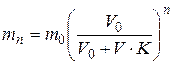 , (6.2)
, (6.2)
де m0 – вихідна кількість розчиненої речовини у об'ємі розчину V0; mn – кількість розчиненої речовини, що залишилась у розчині після n-циклів екстракції; V – об’єм розчинника, що додається до розчину; К – коефіцієнт розподілу.
6.3 Фракційна перегонка
Як відомо, процес кипіння супроводжується утворенням насиченої пари. Залежність складу пари від складу рівноважного з нею розчину двох летких рідин була встановлена Д.П.Коноваловим.
 І закон Коновалова – "Підвищення відносного вмісту компонента в рідкій фазі завжди викликає збільшення його вмісту в парі. При цьому пара порівняно з рівноважною з нею рідиною багатша на той компонент, додавання якого до системи зменшує температуру кипіння суміші". На рис. 6.1 наведена діаграма кипіння системи з двох рідин – А і L. Вище кривої abl, яка показує складнасиченої пари, система знаходиться у газоподібному стані. Нижче кривої adl, яка характеризує складкиплячої рідини, система знаходиться у рідкому стані. Точки a і l відповідають точкам кипіння рідин А і L відповідно. Для визначення складу насиченої пари над киплячою рідкою сумішшю, яка містить по 50 моль. % речовин А і L (точка d) проводимо ізотерму (ноду) до її перетину з кривою abl (точки b) і визначаємо склад пари: 76 % А і 24 % L. Насичена пара, відповідно закону Коновалова, містить більше речовини А, порівняно з рівноважним з нею рідким розчином.
І закон Коновалова – "Підвищення відносного вмісту компонента в рідкій фазі завжди викликає збільшення його вмісту в парі. При цьому пара порівняно з рівноважною з нею рідиною багатша на той компонент, додавання якого до системи зменшує температуру кипіння суміші". На рис. 6.1 наведена діаграма кипіння системи з двох рідин – А і L. Вище кривої abl, яка показує складнасиченої пари, система знаходиться у газоподібному стані. Нижче кривої adl, яка характеризує складкиплячої рідини, система знаходиться у рідкому стані. Точки a і l відповідають точкам кипіння рідин А і L відповідно. Для визначення складу насиченої пари над киплячою рідкою сумішшю, яка містить по 50 моль. % речовин А і L (точка d) проводимо ізотерму (ноду) до її перетину з кривою abl (точки b) і визначаємо склад пари: 76 % А і 24 % L. Насичена пара, відповідно закону Коновалова, містить більше речовини А, порівняно з рівноважним з нею рідким розчином.
Рисунок 6.1 – Діаграма кипіння системи двох летких рідин
ІІ закон Коновалова "На кривих температур кипіння двокомпонентних систем існують екстремальні точки (мах і мін), які відповідають розчинам, склад яких однаковий із складом рівноважної з ним пари. Такі розчини називають азеотропними".
Фракційною перегонкою називають процес розділення розчину на окремі компоненти шляхом багатократного повторення операцій випаровування і конденсації. Фракційну перегонку незручно здійснювати у кілька послідовних стадій, тому її проводять у формі безперервного процесу, при якому операції конденсації і дистиляції автоматизовані. Такий процес називається ректифікацією, а апарати, в яких вона здійснюється – ректифікаційними колонами.
Лекція № 7
Електрохімічні процеси
План лекції
1. Електрохімічні процеси у розчинах електролітів. Іонна сила електролітів.
2. Електроди. Електродний потенціал. Електроди І-го і ІІ-го роду. Окисно-відновні електроди. Рівняння Нернста.
3. Електрохімічні системи. Електрорушійна сила.
4. Вимірювання електродних потенціалів. Водневий електрод.
Рекомендована література: [1] С.409-435, [2] С.76-83, [4] С.182-192, 312-318.
7.1 Електрохімічні процеси у розчинах електролітів
У розчинах електролітів перебігає низка самочинних процесів: дисоціація, сольватація, міжіонна взаємодія і т.д. При цьому дуже важко визначити реальну концентрацію електроліту: кількість іонів у розчині визначається внеском кожного з факторів. Тому замість звичайної концентрації введено поняття "активної концентрації", або активності. Активність – це фізична величина, підстановка якої замість концентрації у будь-які рівняння та закони, що описують властивості розчинів, робить їх придатними для розчинів електролітів. Величина активності відрізняється від значень концентрації на коефіцієнт активності:
a = с×g, (7.1)
де g – коефіцієнт активності, який безпосередньо враховує взаємодію між іонами.
Значення коефіцієнтів активності електролітів залежать від загальної концентрації всіх іонів, присутніх у розчині, та їх валентності. У зв’язку з цим було введене поняття “іонної силирозчину”:
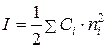 . (7.2)
. (7.2)
Рівняння, за яким визначають коефіцієнти активності електролітів має вигляд:
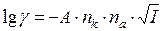 , (7.3)
, (7.3)
де nk і na – заряди катіону і аніону; І – іонна сила електроліту; А – стала, що залежить від природи розчинника, зокрема для води за Т = 298 К А = 0,51.
Електроди
Електродом називають сукупність двох контактуючих фаз, одна з яких є провідником ІІ-го роду (електролітом), а друга – провідником І-го роду, наприклад металом. Метали – це кристалічні речовини, у вузлах кришталевих ґраток яких розташовані катіони металу, а відповідна кількість узагальнених електронів вільно рухається по об’єму металу. За рахунок некомпенсованого обміну катіонами між металом та електролітом на металі виникає заряд. Активні метали звичайно заряджаються негативно, малоактивні – позитивно. За наявності заряду до поверхні металу наближаються іони протилежного знаку, утворюючи на поверхні розділу метал/електроліт подвійний електричний шар іонів. Між електродом і електролітом виникає різниця потенціалів – e, яка вимірюється у вольтах. Величина потенціалу залежить від природи металу і концентрації іонів в електроліті і розраховується за допомогою рівняння Нернста. Усі електроди можна поділити на декілька типів.
Електрод І роду за будовою являє собою метал, занурений в електроліт, що містить катіони цього металу. Електрод І роду можна представити у вигляді схеми: МеlМеn+. На поверхні металу перебігає електродна реакція:
Меn+ + nē ⇄ Me0
Рівняння, за яким визначають потенціали електродів І-го, має такий вигляд:
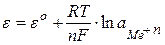 , (7.4)
, (7.4)
де eо – стандартний потенціал електроду, В; n – число електронів, що беруть участь в електродній реакції; F – число Фарадея; a – активність катіонів металу в розчині.
Електрод ІІ роду за будовою являє собою метал, вкритий малорозчинною сполукою цього метала і занурений в електроліт, що містить аніони цієї сполуки. Величина потенціалу електродів ІІ-го роду залежить від активності аніонів, які беруть участь в електродній реакції.
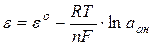 , (7.5)
, (7.5)
де a – активність аніонів у розчині.
Хлорсрібний електрод Ag,AgCllКСl – найбільш поширений з електродів ІІ-го роду. На його поверхні перебігає електрохімічна реакція:
AgCl(т) + ē ⇄ Ag(т) + Cl–.
Окисно-відновний електрод за будовою являє собою пластину інертного металу, занурену в електроліт, що містить окисник і відновник. Потенціал електроду визначається типом ОВР, що перебігає на електроді і розраховується за рівнянням:
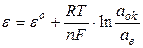 , (7.6)
, (7.6)
де aok – активність речовини-окисника; aв – активність речовини-відновника.
7.3 Електрохімічні системи
Електрохімія – наука, що вивчає взаємне перетворення хімічної і електричної енергії під час перебігу хімічних реакцій в електрохімічних системах. Будь-яка електрохімічна система складається з 3-х основних частин: електроліту; електродів і зовнішнього ланцюгу – металічних провідників, що з’єднують електроди.
Так, елемент Даніеля-Якобі складається з двох металів – міді і цинку, занурених у розчини власних солей. Електроліти розділені мембраною, що не дозволяє електролітам перемішуватися. Схематично елемент записують так:
(–) ZnlZnSO4llCuSO4lCu (+),
a1 a2
де a1 і a2 – активності розчинів ZnSO4 і CuSO4 відповідно.
Цинк, як більш активний метал, придбає більш негативний заряд ніж мідь. Якщо метали з’єднати дротом, то електрони будуть переходити від цинку до міді. Цинк почне розчинятися і переходити у розчин у вигляді іонів, а на мідному електроді будуть розряджатися іони Купруму, що знаходилися в розчині CuSO4. У системі при цьому виникає процес, що складається з двох електродних реакцій:
Zn0 – 2ē = Zn+2;
Cu+2 + 2ē = Cu0.
Різницю електродних потенціалів в елементі називають електрорушійною силою, Е=e+–e–. Рівняння Нернста, за яким визначають ЕРС, має такий вигляд:
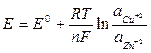 . (7.7)
. (7.7)
де Е0 – стандартна ЕРС елементу, що працює за Т = 298 К і активності іонів електроліту рівних 1 моль/л.
7.4 Вимірювання електродних потенціалів
Визначення потенціалів електродів ґрунтується на вимірюванні різниці потенціалів між двома електродами – індикаторним і електродом порівняння за відсутності струму у зовнішньому ланцюгу. Як електроди порівняння застосовують водневий електрод або електроди ІІ-го роду, потенціали яких є сталими величинами у будь-якому середовищі. Так стандартний електродний потенціал насиченого хлорсрібного електроду дорівнює +0,201 В.
Водневий електрод являє собою платиновий дротик, занурений у розчин кислоти, через який пропускають водень. Схема електроду має такий вигляд – Pt,Н2lН+. На поверхні електроду перебігає оборотна реакція: 2Н+ + 2ē ⇄ Н2.
Рівняння Нернста для водневого електроду має вигляд:
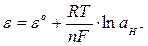 . (7.8)
. (7.8)
Потенціал водневого електроду, що працює за стандартних умов, прийнято рівним нулю. Рівняння Нернста для стандартного водневого електроду має вигляд:
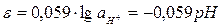 . (7.9)
. (7.9)
Розроблена воднева шкала стандартних електродних потенціалів дозволяє визначати напрямок перебігу електрохімічних процесів. Відповідно цій шкалі потенціал електроду дорівнює ЕРС елементу, в якого один з електродів – стандартний водневий, а інший – електрод, потенціал якого визначається.
Як індикаторні електроди застосовують мембранні електроди, потенціали яких залежать від активності відповідних іонів. Це іон-селективні електроди. Найбільш відомим з них є скляний електрод, який являє собою скляну кульку, виготовлену з електродного скла. Кулька заповнена розчином кислоти з відомою концентрацією іонів Н+, в якому знаходиться контактний півелемент, виготовлений з платини. Величину потенціалу для скляного електроду можна обчислити за формулою:
e = e0 – 0,059рН, (7.10)
Потенціал іонселективного електроду є функцією активності іонів у розчині. Для визначення активності іонів такий електрод з'єднують з електродом порівняння, занурюють електроди у досліджуваний розчин, і вимірюють ЕРС елементу за допомогою потенціометрів, які називають іонометрами. Шкала цих приладів показує логарифм активності іонів, концентрацію яких визначають.
Лекція №8
Основні положення хімічної кінетики
План лекції
1. Швидкість хімічних реакцій. Закон діючих мас. Константа швидкості.
Правило Вант-Гоффа. Аналіз рівняння Арреніуса.
2. Кінетична класифікація хімічних реакцій. Молекулярність і порядок реакції. Кінетичні рівняння хімічних реакцій І-го та ІІ-го порядків.
3. Методи визначення порядків хімічних реакції.
Рекомендована література: [1] С.456-464, [2] С.68-71, [3] С.61-67,
[4] С.121-126.
8.1 Швидкість хімічних реакцій
Хімічна кінетика – розділ фізичної хімії, що вивчає швидкість хімічних реакцій та механізм їх перебігу. Швидкість хімічної реакції визначається за зміною концентрації реагентів або продуктів реакції за одиницю часу. Швидкість хімічної реакції залежить: від концентрації реагентів; умов перебігу реакції; природи реагентів; наявності каталізаторів або інгібіторів.
Закон діючих мас. Швидкість хімічних реакцій пропорційна числу зіткнень, яких зазнають молекули реагентів під час реакції, тобто від концентрації реагентів. Ця залежність одержала назву закону дії мас: "Швидкість хімічної реакції пропорційна добутку концентрацій реагуючих речовин, зведених у степені їх стехіометричних коефіцієнтів". Для абстрактної реакції:
aA + bB → dD + rR (8.1)
швидкість хімічної реакції розраховують за рівнянням:
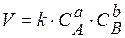 , (8.2)
, (8.2)
де СА і СВ – концентрації вихідних речовин; k – константа швидкості реакції.
Константа швидкості хімічної реакції – це фізична величина, яка дорівнює швидкості реакції, за умови, що концентрації усіх реагентів дорівнюють 1 моль/л. Константа швидкості реакції – основна кінетична характеристика реакцій, оскільки вона не залежить від концентрації реагентів.
Правило Вант-Гоффахарактеризує залежність швидкості реакцій від температури: "При підвищенні температури на кожні 10 градусів швидкість реакцій зростає у 2...4 рази". Правило Вант-Гоффа носить виключно емпіричний характер і виконується в інтервалі температур 0...1000 С. Теоретичну залежність швидкості хімічної реакції від температури виражає рівняння Арреніуса:
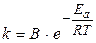 , (8.3)
, (8.3)
де k – константа швидкості реакції; B ‑ коефіцієнт пропорційності; Еa ‑ енергія активації, Дж; R ‑ газова стала, 8,31 Джмоль×К; Т ‑ температура, К.
Енергія активації – це мінімальна енергія, яку необхідно надати молекулам, щоб зробити їх активними, тобто здатними вступати в хімічну реакцію. Чим менша енергія активації, тим більше швидкість хімічної реакції.
8.2 Кінетична класифікація хімічних реакцій
У хімічних реакціях можуть брати участь іони, радикали або молекули. Відповідно до цього за механізмом перебігу розрізняють іонні, радикальні та молекулярні реакції. Молекулярні реакції у залежності від кількості молекул, що приймають участь в елементарному акті хімічної взаємодії, поділяються за молекулярністю на моно-, бі-, і тримолекулярні. Реакції, в яких беруть участь більш ніж три часточки, – це складні реакції, що складаються з декілька стадій.
Сума показників ступенів при концентраціях у кінетичному рівнянні реакції визначає порядок реакції. Прості хімічні реакції можуть бути І-го, ІІ-го та ІІІ-го порядків, у складних реакції порядок може бути величиною дробовою. Кінетичні рівняння хімічних реакцій І-го і ІІ-го порядку мають відповідно такий вигляд:
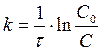 ; (8.4)
; (8.4)
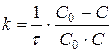 , (8.5)
, (8.5)
де С0 – вихідна концентрація речовини; С – її концентрація на момент часу t.
Існує ще одна кінетична характеристика хімічних реакцій: період піврозпаду – час, за який реагує половина вихідної кількості реагенту.
8.3 Методи визначення порядку хімічних реакцій
Метод надлишку. Для визначення порядку реакції спочатку встановлюють порядок реакції за кожним реагентом. Для визначення порядку за окремим реагентом концентрації інших реагентів повинні бути настільки великими, щоб їх зміною з часом можна було знехтувати. Так, для реакції (8.1.) порядок реакції n дорівнює сумі a + b. Якщо речовину А беруть у великому надлишку, швидкість хімічної реакції буде залежати тільки від концентрації речовини В:
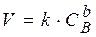 . (8.6)
. (8.6)
Якщо ж у надлишку узяти речовину В, то закон дії мас має такий вигляд:
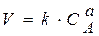 . (8.7)
. (8.7)
Далі, користуючись методом підстановки, знаходять коефіцієнти а і b.
Метод підстановки. Суть методу полягає в тому, що через певні проміжки часу визначають концентрацію реагенту або продукту реакції. Визначені величини концентрацій послідовно підставляють у кінетичні рівняння для реакцій І-го, ІІ-го або ІІІ-го порядку. Рівняння, за яким одержали сталі значення константи швидкості, описує кінетику досліджуваної реакції. Якщо ж рівняння не призвели до одержання сталих значень константи швидкості, то досліджувана реакція є складною.
Метод Вант-Гоффа дозволяє визначати як цілі, так і дробові порядки. Для визначення порядку реакції за методом Вант-Гоффа необхідно знати швидкість реакції за 2-х концентраціях реагенту. Порядок реакції визначають за формулою:
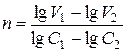 , (8.8)
, (8.8)
де V1 i V2 – швидкість реакції за концентрацій реагенту С1 і С2 відповідно.
Лекція №9
Адсорбційні процеси на поверхні
План лекції
1. Поверхневий натяг рідин. Методи вимірювання поверхневого натягу.
2. Поверхнево-активні речовини (ПАР). Ізотерма поверхневого натягу.
Поверхнева активність речовин. Правило Дюкло-Траубе.
3. Адсорбція ПАР на межі поділу розчин-газ. Рівняння адсорбції Гіббса.
4. Теорія фізичної адсорбції Ленгмюра. Рівняння Фрейндліха. Хемосорбція.
5. Поверхневі явища. Адгезія і когезія. Розтікання рідин. Змочування. Рівняння Юнга.
6. Сорбенти. Класифікація сорбентів. Гідрофобні та гідрофільні сорбенти.
Рекомендована література: [1] С.348-391, [2] С.85-99, [3] С. 51-55, 105-118.
9.1 Поверхневий натяг рідин
Поверхневий шар рідин за своїми властивостями суттєво відрізняється від її внутрішніх шарів. Кожна молекула усередині рідини притягує до себе навколишні молекули й одночасно з такою самою силою притягується рівномірно молекулами, що оточують її. Рівнодіюча сил міжмолекулярної взаємодії при цьому дорівнює нулю. В іншому стані перебувають молекули, що знаходяться у поверхневому шарі рідини. На них діють сили притягання тільки від молекул нижньої півсфери. Дією молекул газу, що знаходяться над поверхнею рідини, можна знехтувати. Рівнодійна міжмолекулярних сил у цьому разі направлена нормально до поверхні усередину рідини. Тобто, молекули на поверхні рідини завжди перебувають під дією сили, яка намагається втягнути їх усередину.
Сила, що діє на одиницю довжини межі поділу називається поверхневим натягом або питомою вільною поверхневою енергією – s. Вимірюється поверхневий натяг у Дж/м2 або Н/м. Для вимірювання поверхневого натягу рідин існує безліч експериментальних методів, найпростіший з них – сталагмометричний. Цей метод ґрунтується на визначенні маси краплі, що відривається від нижнього кінця капіляру, заповненого досліджуваною рідиною. Чим більша маса краплі рідини, тим більший поверхневий натяг має ця рідина.
9.2 Поверхнево-активні речовини
Поверхня розділу фаз у гетерогенних системах має надлишок поверхневої енергії за рахунок неврівноважених міжмолекулярних зв’язків. Це дозволяє адсорбуватися (накопичуватися) на межі поділу фаз поверхнево-активним речовинам (ПАР), поверхневий натяг при цьому знижується. Молекули ПАР мають дифільну будову і складаються з неполярного вуглеводневого радикалу і полярної гідрофільної функціональної групи. Вони поділяються на три групи.
Аніоноактивні ПАР, які дисоціюють у воді з утворенням поверхнево-активного аніону. До ПАР цього типу відносяться, наприклад, вищі карбонові кислоти та їх солі (мила) загальної формули R--СООМе, де Ме – лужний метал.
Катіоноактивні ПАР – сполуки, якідисоціюють у воді з утворенням поверхнево-активного катіону. До катіоноактивних ПАР відносять солі первинних, вторинних і третинних аліфатичних амінів.
Амфолітні ПАР, які містять дві функціональні групи, одна з яких носить кислотний, а інша основний характер. До амфолітних ПАР відносять амінокислоти містять карбоксильну та амонійну групи. У залежності від рН середовища вони можуть виявляти властивості як аніоноактивних, так і катіоноактивних ПАР.
Неіоногенні ПАР – сполуки, які під час розчинення не дисоціюють на іони, а поверхнево-активні властивості виявляє уся молекула ПАР. Молекули таких ПАР складаються з вуглеводневого радикалу, на кінці якого розміщено декілька неіоногенних гідрофільних груп, наприклад, гідроксильних або етерних.
Для визначення поверхневого натягу розчинів ПАР користуються емпіричним рівнянням Шишковського:
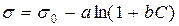 , (9.1)
, (9.1)
де s і s0 – поверхневий натяг розчину і розчинника відповідно; С – концентрація розчину ПАР; а і б – емпіричні коефіцієнти.
Графічна залежність поверхневого натягу розчину ПАР від її концентрації приведена на рис.9.1. Зміна поверхневого натягу розчину від концентрації ПАР – ¶s/¶С є мірою її поверхневої активності. Для визначення поверхневої активності речовини проводять дотичну до кривої "s–С" і розраховують тангенс кута нахилу: tga = ¶s/¶С.
 Поверхнева активність ПАР залежить від будови їх молекул. Так, у свій час Дюкло і Траубе встановили, що у гомологічному ряду органічних кислот і спиртів подовження вуглеводневого радикалу на кожну групу СН2 призводить до збільшення поверхневої активності у 3,0-3,5 рази.
Поверхнева активність ПАР залежить від будови їх молекул. Так, у свій час Дюкло і Траубе встановили, що у гомологічному ряду органічних кислот і спиртів подовження вуглеводневого радикалу на кожну групу СН2 призводить до збільшення поверхневої активності у 3,0-3,5 рази.
Рисунок 9.1 – Ізотерма поверхне-вого натягу водного розчину ПАР
9.3 Адсорбція ПАР на поверхні рідин
Адсорбція – це самочинний процес накопичення речовини на поверхні рідини. Адсорбція ПАР відбувається у поверхневому шарі рідини, який називають адсорбційним. Вивчення будови адсорбційних шарів показало, що вони складаються з одного ряду молекул, орієнтованих певним чином. У полярних розчинниках, таких як вода, функціональні групи розміщуються на поверхні розчину, а вуглеводневі радикали повернені в бік повітря. У неполярних розчинниках орієнтація молекул ПАР протилежна: радикали повернені в бік рідини, а функціональні групи – у бік повітря. Адсорбція ПАР на поверхні рідин розраховується за термодинамічним рівнянням Гіббса:
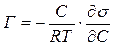 ,(9.2.)
,(9.2.)
де Г – адсорбція, моль/м2; С – концентрація розчину ПАР, моль; R – універсальна газова стала; Т – температура, К; ds/dC – поверхнева активність.
9.4 Теорія фізичної адсорбції Ленгмюра
Поверхня твердих тіл має надлишок поверхневої енергії за рахунок неврівноважених зв’язків у кристалічних ґратах, що дозволяє їм адсорбувати речовини з навколишнього середовища. Речовини, що здатні адсорбувати інші речовини, називають сорбентами. На величину адсорбції впливають природа і геометрія поверхні сорбенту. Вимірюють адсорбцію у моль/м2 або у моль/г.
У 1916 році І.Ленгмюр сформулював теорію фізичної адсорбції:
· адсорбція молекул відбувається тільки на активних центрах сорбенту, як правило на виступах поверхні;
· на кожному активному центрі адсорбується тільки одна молекула;
· адсорбція відбувається за рахунок міжмолекулярних сил взаємодії;
· процес адсорбції оборотний: молекули адсорбтиву періодично відриваються від поверхні сорбенту, а їх місце займають інші молекули;
· взаємодія між сусідніми адсорбованими молекулами відсутня.
На основі цих положень Ленгмюр аналітично вивів рівняння, яке має вигляд:
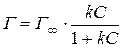 , (9.3)
, (9.3)
 де Г¥ – максимальна адсорбція, відповідає моменту, коли на поверхні сорбенту утворюється мономолекулярний шар адсорбтиву; k – стала Ленгмюра. Залежність величини адсорбції від концентрації розчину приведена на рис. 9.2.
де Г¥ – максимальна адсорбція, відповідає моменту, коли на поверхні сорбенту утворюється мономолекулярний шар адсорбтиву; k – стала Ленгмюра. Залежність величини адсорбції від концентрації розчину приведена на рис. 9.2.
Рисунок 9.2 – Ізотерма адсорбції Ленгмюра
Теорія Ленгмюра не враховує можливість взаємодії між молекулами адсорбтиву. Пізніше була створена теорія БЕТ, яка врахувала цю взаємодію.
У випадках, коли сорбент являє собою порошок або пористе тіло, величину адсорбції звичайно визначають за емпіричним рівнянням Фрейндліха:
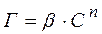 , (9.4)
, (9.4)
де Г – адсорбція; С – рівноважна молярна концентрація адсорбтиву у розчині; b і n –емпіричні коефіцієнти, сталі для кожної пари адсорбтив-сорбент.
Якщо адсорбтив і матеріал сорбенту мають високу хімічну спорідненість, то стає можливою хімічна взаємодія між ними. Таке явище називають хемосорбцією.
Хемосорбція процес екзотермічний. специфічний і необоротний, який з підвищенням температури посилюється. При хемосорбції на поверхні сорбенту утворюються продукти хімічної реакції між адсорбтивом і сорбентом.
9.5 Поверхневі явища
У гетерогенних системах розрізняють міжмолекулярну взаємодію, що відбувається в межах однієї фази і взаємодію між окремими фазами. Взаємодія атомів і молекул в межах однієї фази називають когезією або зчепленням. Найбільша когезія має місце в твердих тілах, найменша – в газах. Кількісно когезія визначається енергією, що витрачається на розрив тіла по площині рівній 1 м2. Через те, що при розриві твердого тіла утворюються дві нові поверхні, площа яких складає дві одиниці, робота когезії дорівнює подвоєному значенню поверхневого натягу на межі тверде тіло-газ: Wк = 2s.
Міжмолекулярна взаємодія між двома поверхнями різних тіл, що приведені в контакт називається адгезією або здатністю прилипати. Адгезія буває трьох типів: між твердими поверхнями (Т-Т); між твердою і рідкою поверхнею (Т-Р); між рідкими поверхнями (Р-Р). Адгезія – це самочинний процес. Величина адгезії визначається енергією, що витрачається на подолання сил зчеплення між двома контактуючими поверхнями. Найбільшу адгезію мають фарби, клеї, бетон то що. Розраховують величину адгезії за рівнянням Дюпре:
Wа = sрг + sтг – sтр (9.5)
Співвідношення між роботою когезії та роботою адгезії характеризує таке явище, як розтікання. Явище розтікання має місце при контакті двох рідин. Припустимо, що на поверхню води нанесли краплю якоїсь рідин. Якщо сили притягання між молекулами рідини (когезія) більші, ніж сили притягання між молекулами рідини і води (адгезія), то рідина збереже форму краплі і розтікання не буде. Навпаки, якщо когезія рідини менша за її адгезію по відношенню до води, відбудеться розтікання рідини по поверхні води. Критерієм розтікання є різниця між Wк і Wа. Цю величину називають коефіцієнтом розтікання.
Wр = Wа – Wк (9.6)
На практиці відомо, що рідина, поверхневий натяг якої менший, розтікається по поверхні тієї рідини, поверхневий натяг якої більше. Як правило, неполярні рідини добре розтікаються по поверхні більш полярної. Змочування – це сукупність явищ, що виникають під час контакту твердого тіла та рідини. Якщо краплину рідини нанести на тверду поверхню, то можна спостерігати такі випадки: краплина розтікається по поверхні тонким шаром; краплина частково розтікається по поверхні, утворюючи з нею певний крайовий кут q (рис. 9.3 а); краплина залишається на поверхні у вигляді кульки (рис. 9.3 б). Змочування твердого тіла рідиною має місце у випадку, коли краплина рідини розтікається на його поверхні.
Критерієм, за яким визначають здатність поверхні змочуватися рідиною, є косинус крайового кута cosq. Якщо cosq >0 – рідина змочує тверду поверхню; якщо cosq < 0 – змочування не відбувається. Визначають cosq за рівнянням Юнга:
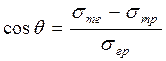 , (9.7)
, (9.7)
 де sтр,sтг,sрг – поверхневі натяги на межах поділу "тверде тіло-рідина"; "тверде тіло-газ" і "рідина-газ" відповідно.
де sтр,sтг,sрг – поверхневі натяги на межах поділу "тверде тіло-рідина"; "тверде тіло-газ" і "рідина-газ" відповідно.
Рисунок 9.3 – Змочування поверхні твердого тіла краплиною рідини.
Вода і водні розчини змочують речовини з іонним або полярним ковалентним зв’язками. Поверхню таких речовин називають гідрофільною. Речовини, такі як парафін або жири, поверхня яких не змочується водою відносять до гідрофобних. Чим краще розчинник змочує сорбент, тим гірша адсорбція розчинених речовин з цього розчинника. Так, активоване вугілля, яке не змочується водою, є найкращим сорбентом для водних розчинів.
На явищі змочування ґрунтується процес флотації – метод розділення суміші гідрофільного і гідрофобного порошків, який ґрунтується на вибірковому змочуванні їх різними рідинами. Флотація буває двох типів: масляна і пінна.
Сорбенти
Сучасна класифікація поділяє сорбенти на мінеральні, вуглецеві і полімерні. До мінеральних відносяться алюмосилікати (цеоліти, бентоніти) і силікагель, який одержують осадженням полімерної силікатної кислоти з розчинів силікатів. Змінюючи умови осадження і висушування одержаного гелю одержують силікагелі різної пористості. На поверхні силікагелю розташована велика кількість функціональних груп ºSi–ОН, завдяки чому він добре адсорбує воду та інші полярні речовини, які здатні утворювати водневі зв’язки.
Найбільш поширеним вуглецевим сорбентом є активоване вугілля. Одержують його з кам’яного або деревного вугілля. Під час випалювання вугілля у присутності води вигоряють смолисті речовини; при цьому усередині матеріалу розвивається пористість і збільшується питома поверхня. Активоване вугілля добре адсорбує неполярні органічні речовини і застосовується для освітлення розчинів і очистки повітря.
Серед полімерних сорбентів на цей час найбільш поширені іонообмінні смоли – іоніти. Іоніти – органічні полімери, що містять активні іоногені групи. Іоніти мають матричну структуру, до складу якої входять фіксовані іони. Ці іони здатні під час контакту з розчинами електролітів обмінюватися на катіони або аніони розчинених речовин. Якщо фіксованими іонами є катіони, то іоніт виконує роль катіоніту, якщо ж матриця має негативний заряд, то такий іоніт є аніонітом. Іоніти застосовують для очистки харчових продуктів і води від надлишкових іонів.
Рекомендована література
1. Киреев, В. А. Курс физической химии [Текст] / В. А. Киреев. – 5-е изд. – М. : Высшая школа, 1985. – 620 с.
2. Савгіра, Ю. О. Фізична та колоїдна хімія [Текст]: Навчальний посібник / Ю.О.Савгіра, С. О. Самойленко, О. В. Добровольська, О. Ф. Аксенова. – Харків : ХДУХТ, 2006.– 162 с.
3. Ліпатніков, В. Є. Фізична та колоїдна хімія [Текст] / В. Є. Ліпатніков, К.М.Казаков. – Київ: Вища школа, 1993. – 197 с.
4. Романова, Н. В. Загальна та неорганічна хімія [Текст] / Н. В. Романова. – Київ: Ірпинь, 1998. – 480 с.
Розділ 3. ОРГАНІЧНА ХІМІЯ
ПРЕДМЕТ ОРГАНІЧНОЇ ХМІЇ. ОСНОВНІ ПОНЯТТЯ ОРГАНЧНОЇ ХІМІЇ
Лекція №1 (2 години)
План
1. Предмет органічної хімії.
2. Розвиток теоретичних уявлень в органічній хімії.
3. Хімічний зв’язок в органічних сполуках.
4. Класифікація органічних сполук та реакцій за напрямком та механізмом.
5. Номенклатура органічних сполук.
1. Предмет органічної хімії
На початку XIX ст. поняття «органічна хімія» використовувалося стосовно хімії сполук, із яких складаються рослинні та тваринні організми. Виникнення теорії віталізму (від латинського vitalis – життєвий).
Органічна хімія – хімія вуглеводнів та їх похідних, до складу яких можуть входити майже всі елементи періодичної системи.
Значення органічної хімії. Вивчення основних теоретичних положень органічної хімії сприяє формуванню, розширенню і поглибленню фундаментальних, загальнопрофесійних, спеціальних знань, а також активному формуванню предметних і професійних компетенцій, спрямованих на виконання виробничих функцій.
2. Розвиток теоретичних уявлень в органічній хімії
Теорія радикалів (20‑30 рр. XIX ст. — Гей-Люсак, Велер, Лібіх).
Теорія типів (40‑50 рр. XIX ст. — Жерар, Дюма, Кекуле).
Теорія хімічної будови органічних сполук О.М. Бутлерова (1861 р.) та її значення. Суть теорії будови можна сформувати у вигляді наступних положень:
1. Атоми в молекулах розташовані не безладно. У молекулах існує певний порядок зв’язку атомів, який називають хімічною будовою (або просто “будовою”).
2. Властивості речовин залежать не лише від того, які атоми та в якій кількості входять до складу молекул, а й від того, в якому порядку вони сполучені між собою, тобто від хімічної будови молекул.
3. Атоми або групи атомів у молекулах взаємно впливають один на одного безпосередньо або через інші атоми. Найбільше впливають безпосередньо зв’язані атоми. Взаємний вплив атомів, безпосередньо не зв’язаних, значно слабший.
4. Відмінність у будові за однакового складу та молекулярної маси зумовлює явище ізомерії.
5. Хімічну будову можна встановити, вивчаючи властивості та перетворення речовин, і виразити її певною формулою.
Теорія О.М.Бутлерова не тільки пояснювала будову молекул усіх відомих органічних сполук та їх властивості, а й дала можливість теоретично передбачувати існування невідомих і нових сполук, знаходити шляхи їх синтезу. Подальшим розвитком теорії будови стало вчення про просторову будову молекул органічних сполук.
Розвиток теорії хімічної будови, стереохімічна гіпотеза Вант-Гофа та Ле-Беля. Гіпотеза О.М. Бутлерова (1862 р.) про тетраедричну будову атому Карбону.
3. Хімічний зв'язок в органічних сполуках
Типи хімічних зв'язків на ( іонні, ковалентні, водневі, металічні й вандерваальсівські).
В молекулах органічних сполук атомами сполучаються, як правило, ковалентний зв’язок. Ковалентний зв’язок характеризується енергією, довжиною, полярністю і поляризованістю зв’язку та просторовою направленістю.
Квантово-механічні уявлення про природу ковалентного зв’язку. Електронна будова атому Карбону. Поняття про гібридизацію. Електронна будова простих, подвійних та потрійних карбон-карбонових зв’язків, s- та p- зв’язки.
Для атома Карбону розрізняють три валентні стани, які визначаються типом гібридизації.
Перший валентний стан – 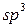 -гібридизація. У цьому випадку утворюються чотири гібридних орбіталі за рахунок гібридизації
-гібридизація. У цьому випадку утворюються чотири гібридних орбіталі за рахунок гібридизації 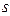 -орбіталі із трьома
-орбіталі із трьома 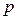 -орбіталями, які між собою утворюють кут 109°28¢ і орієнтовані до вершин тетраедра. Ці орбіталі приймають участь в утворенін s- зв’язків.
-орбіталями, які між собою утворюють кут 109°28¢ і орієнтовані до вершин тетраедра. Ці орбіталі приймають участь в утворенін s- зв’язків.
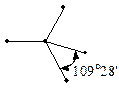
|
Другий валентний стан атому Карбону – 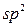 -гібридизація, за умов якої відбувається гібридизація однієї
-гібридизація, за умов якої відбувається гібридизація однієї 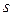 - і двох -орбіталей. Утворюються три однакові гібридизовані орбіталі, які розміщені в одній площині під кутом 120°. Негібридизована -орбіталь розташована перпендикулярно до цієї площини. Така гібридизація характерна для сполук із подвійним зв’язком, наприклад етену
- і двох -орбіталей. Утворюються три однакові гібридизовані орбіталі, які розміщені в одній площині під кутом 120°. Негібридизована -орбіталь розташована перпендикулярно до цієї площини. Така гібридизація характерна для сполук із подвійним зв’язком, наприклад етену 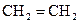 . Подвійний зв'язок є сполученням , s- та p- зв’язків.
. Подвійний зв'язок є сполученням , s- та p- зв’язків.
Третій валентний стан атому Карбону – 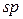 -гібридизація. Цей тип гібридизації здійснюється внаслідок гібридизації однієї - і однієї -орбіталі. Гібридизовані орбіталі розміщені лінійно під кутом 180°. Дві негібридизовані -орбіталі розташовані у взаємно перпендикулярних площинах і, перекриваючись бічними поверхнями, утворюють два p-зв’язки. Потрійний зв’язок являє собою сполучення одного s- і двох p-зв’язків. Така гібридизація характерна для сполук із потрійним зв’язком, наприклад етину
-гібридизація. Цей тип гібридизації здійснюється внаслідок гібридизації однієї - і однієї -орбіталі. Гібридизовані орбіталі розміщені лінійно під кутом 180°. Дві негібридизовані -орбіталі розташовані у взаємно перпендикулярних площинах і, перекриваючись бічними поверхнями, утворюють два p-зв’язки. Потрійний зв’язок являє собою сполучення одного s- і двох p-зв’язків. Така гібридизація характерна для сполук із потрійним зв’язком, наприклад етину 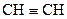 .
.
4. Класифікація органічних сполук та реакцій за механізмом та
Напрямком
В основу класифікації положено уявлення про будову карбонового ланцюгу молекули.

Для кожного із наведених класів органічних сполук прийнято окрему класифікацію залежно від наявності тих або інших функціональних груп, які визначають їх хімічні властивості.
Класифікація реакцій органічних сполук за механізмом: радикальні (R) та іонні (N, E); за напрямком та результатом реакції: заміщення — S (substitution);
приєднання — A (addition); відщеплення, елімінування — E (elimination).
Дата добавления: 2016-12-16; просмотров: 2507;