Основные положения теории сильных электролитов.
| |
Исходя из того, что сильные электролиты полностью диссоциированы, можно было ожидать, что коэффициент Вант-Гоффа i для электролита, диссоциирующего, например, на два иона, должен равняться двум не только в разбавленных, но и в достаточно концентрированных растворах. Однако опыты не подтверждают этого. Коэффициент i в растворах сильных электролитов в значительной степени зависит от концентрации электролита, уменьшаясь с увеличением концентрации раствора. Такая зависимость i от концентрации в растворах объясняется взаимодействием ионов между собой.
В электрическом поле постоянного тока ионы в растворах сильных электролитов имеют меньшую подвижность ввиду межионного взаимодействия.
Дело в том, что под влиянием внешнего электрического поля «ионная атмосфера» смещается к одному полюсу, а ион, находящийся в центре этой атмосферы, стремится к другому полюсу. Кроме того, увлекаемые ионами сольватные (гидратные) оболочки также способствуют их торможению. Чем выше концентрация растворов, тем плотнее «ионная атмосфера» и тем медленнее движутся ионы.
Межионное взаимодействие, а также сольватация ионов уменьшают не только абсолютную скорость их движения, но и осмотическое давление растворов, величину понижения давления пара над ними и т. д. Создается впечатление, что в растворе находится меньше ионов, чем на самом деле. Поэтому величина а является не истинной, а кажущейся степенью электролитической диссоциации сильного электролита.
В слабых электролитах, растворы которых содержат относительно малое количество ионов, взаимодействие последних сравнительно невелико. Кажущаяся степень диссоциации для них практически отвечает истинному значению.
Как известно, уравнения Генри, Вант-Гоффа, Рауля и другие термодинамические уравнения хорошо описывают свойства предельно разбавленных (идеальных) растворов. Свойства растворов сильных электролитов, рассчитанные по этим уравнениям, значительно отличались от фактически наблюдаемых. Многочисленные исследования показали, что введение в эти и другие термодинамические уравнения поправочных коэффициентов, учитывающих отклонение от идеального состояния, нецелесообразно, так как при этом сами уравнения становятся чрезмерно сложными.
Поэтому дальнейшее развитие теории растворов пошло по другому пути. Форма уравнений, описывающих свойства идеальных растворов, была сохранена неизменной, но применимость этих уравнений к реальным системам достигалась тем, что в них вместо обычных величин, характеризующих системы (давления, концентрации), стали использовать величины, заимствованные из опыта. В настоящее время все термодинамические расчеты свойств растворов сильных электролитов строятся на использовании введенной Льюисом величины активности электролита, или активности его ионов. Активность определяется как величина, подстановка которой вместо концентрации в термодинамические уравнения, действительные для простейших (идеальных) систем, делает их применимыми к рассматриваемым растворам.
Активности отличаются от концентраций только тем, что в них входят силы взаимодействия, существующие в растворах и не зависящие от природы растворенных частиц, а также от их концентрации. Поэтому активность можно представить как произведение концентрации на некоторый переменный фактор, называемый коэффициентом активности, т. е.

| 4.22 |
где а — активность электролита (или его ионов); С — аналитическая концентрация электролита; f — коэффициент активности, включающий поправку на силы взаимодействия.
Коэффициенты активности можно найти, сравнивая аналитические концентрации с теми величинами, которые следует подставить в уравнение для растворов электролитов, чтобы получить полное соответствие с данными опыта. Обычно их определяют экспериментально по величине осмотического давления, по понижению температуры замерзания, по повышению температуры кипения раствора или же измерением э. д. с. соответствущей гальванической цепи.
Коэффициент активности, как правило, бывает меньше единицы; коэффициент активности становится равным единице лишь при очень большом разбавлении раствора, когда силы взаимодействия между ионами приближаются к нулю. В этом случае а ~ С, т. е. движение ионов в растворе не стеснено. У сильных электролитов, например, это имеет место только в очень разбавленных растворах при С < 0,0001 кмоль/м3. В таких растворах расстояние между ионами достаточно большое, и межионные силы не оказывают влияния на скорость их передвижения.
Если коэффициент активности меньше единицы, активность ионов меньше их концентрации, получившейся при диссоциации растворенного вещества: а < С. Так, в 0,1 М растворе соляной кислоты активная концентрация Н+ и Сl- получается равной всего СHCl·2f≈0,1·2·0,814 = 0,163 кмоль/м3 вместо 0,1·2 = 0,2 кмоль/м3, так как коэффициент активности f для однозарядных ионов Н+ и Сl- в 0,1 М растворе НСl равен 0,814. Активность же ионов Н+ в 0,1 М растворе соляной кислоты

Необходимо отметить, что при очень больших концентрациях некоторых электролитов f вновь начинает расти, что объясняется недостатком молекул воды для гидратации всех ионов. Ионы, частично или полностью лишенные гидратной оболочки, особенно легко подвижны. Активность в подобных случаях оказывается выше действительной концентрации частиц, а коэффициент активности становится больше единицы.
В водных растворах коэффициент активности данного электролита (или данного иона) зависит в основном от концентраций и валентностей всех присутствующих ионов. Коэффициент активности того или иного вещества может быть определен экспериментально различными методами. Необходимо отметить только, что величину коэффициентов активности отдельных ионов опытным путем определить нельзя, так как всегда результат получается итоговый для растворенного вещества в целом.
Для характеристики зависимости активности иона от концентрации всех находящихся в растворе ионов Льюис ввел понятие об ионной силе раствора электролита. Ионной силой раствора электролита называется величина (µ), измеряемая полусуммой произведения концентрации (С) каждого из присутствующих в растворе ионов на квадрат их валентности (z), т. е.
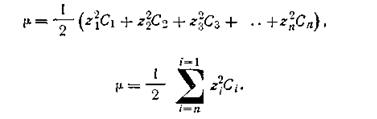
| 4.23 |
Недиссоциированные молекулы, как не имеющие зарядов, в формулу подсчета ионной силы раствора не включаются.
Приведем пример вычисления ионной силы раствора для 0,4 М раствора Na2SO4. Для решения используем формулу:

| 4.24 |
С увеличением концентрации раствора сильного электролита количество ионов в растворе возрастает, что приводит к увеличению ионной силы раствора и значительному уменьшению коэффициента активности, а следовательно, и активности всех ионов.
Для разбавленных растворов, ионная сила которых не превышает 0,01, коэффициент активности ионов связан с ионной силой раствора следующим соотношением:
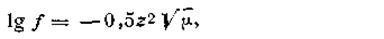
| 4.25 |
где z — заряд иона, а µ—ионная сила раствора.
Из этой формулы следует, что чем больше ионная сила раствора, тем меньше коэффициент активности его ионов; если ионные силы двух растворов равны, то коэффициенты активности равновалентных ионов также одинаковы. В табл. 14 приведены средние значения коэффициентов активности ионов в зависимости от величины ионной силы раствора.
Коэффициент активности f широко используется в практике и в теоретических расчетах. Активность ионов (выраженная, как и концентрация, в кмоль/м3) является на самом деле эффективной концентрацией, проявляющей себя при химических реакциях. Обычные же концентрации показывают количество вещества, находящееся в растворе. Поэтому при теоретических расчетах химических реакций в растворах в связи, например, с использованием закона действующих масс необходимо брать не концентрации веществ, находящихся в растворе, а действующие активные массы исходных реагентов и образующихся продуктов реакции, которые в данный момент непосредственно участвуют в химическом процессе.
Теория сильных электролитов, развитая Дебаем и Гюккелем, при большой сложности математического аппарата применима только при концентрациях, не превышающих 0,01—0,05 н. Выводы этой теории хорошо согласуются с экспериментальными данными для очень разбавленных водных растворов. Для более высоких концентраций она оказывается непригодной, чтобы достаточно полно охарактеризовать чрезвычайно сложную картину взаимодействия между частицами, находящимися в растворе.
Плотность ионной атмосферы, ее радиус, скорость возникновения и разрушения сложным образом влияют на термодинамические и электропроводные свойства электролита. Количественно учесть влияние всех этих фактов теория Дебая и Гюккеля была в состоянии только для простейших электролитов и при условии очень сильного разбавления.
В настоящее время установлено, что в более концентрированных растворах между заряженными ионами возникает взаимодействие не только электростатического, но и химического порядка. В частности, было установлено, что в концентрированных растворах электролитов в воде (а в неводных растворителях с низкой диэлектрической постоянной и при умеренных концентрациях электролита) возможно образование ионных пар, или ионных двойников. Ионные двойники из положительно и отрицательно заряженных ионов появляются в результате действия чисто кулоновских сил, поэтому они менее прочны, чем недиссоциированные молекулы электролита. Однако связи, удерживающие ионы вместе, достаточно сильны для того, чтобы первоначальные ионы потеряли свою самостоятельность и стали проявлять свойства незаряженных частиц.
Например, образование ионных двойников из исходных индивидуальных ионов при растворении хлорида калия можно схематически представить следующим образом: 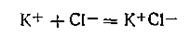
Такая ассоциация КСl в воде возможна при концентрации 27 кмоль/м3.
Более поздними исследованиями было установлено, что в концентрированных растворах кроме незаряженных ионных двойников можно ожидать также образования ионных тройников, в которых заряды ионов не уничтожаются:
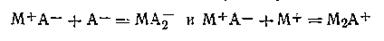
Образование ионных тройников можно представить себе также как результат ассоциации двух ионных пар с последующей ионизацией возникших комплексов:
и 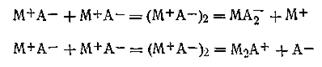
В настоящее время считается экспериментально доказанным, что наряду с заряженными ионными тройниками в концентрированных растворах электролитов (особенно в неводных растворах) могут присутствовать также незаряженные ассоциированные соединения. Эти соединения могут образоваться по схеме

Образование ионных двойников, тройников и незаряженных комплексов является причиной особого поведения сильных электролитов в неводных средах: уменьшения изотонического коэффициента, снижения осмотического давления, электрической проводимости и т. д. по сравнению с водными растворами равнозначных концентраций.
Таким образом, современная теория растворов сильных электролитов, развитая на основе представлений Дебая и Гюккеля, еще далека от совершенства. Вывод теоретических уравнений для расчета коэффициентов активности, применимых в широкой области концентраций, требует создания более точных представлений о молекулярном строении электролитов и о природе тех сил, которые наряду с кулоновскими силами и силами теплового движения действуют между всеми частицами раствора.
Дата добавления: 2016-02-20; просмотров: 2908;