Методи досліджень
Методи органолептичної оцінки
Органолептичний метод — це метод визначення якості продукції безпосередньо за допомогою органів відчуттів людини: (зору, слуху, дотику, смаку, запаху).
Органолептична оцінка товару — узагальнення оцінки його якості, здійснений лише за допомогою органів відчуттів людини. Оцінюються як зовнішні характеристики такі як вигляд, колір, форма, прозорість, запах так і такі як смак, м’якість тощо. Часто використовується для оцінювання напоїв: алкогольних напоїв, вина, кави, чаю, а також кондитерських виробів. Часто допомагає зрозуміти міру свіжості сировини, дотримання технології процесів виробництва чи вирощування певного продукту.
Різновидом органолептичного методу є сенсорний, дегустаційний та ін методи. Сенсорний аналіз застосовується для оцінки якості продуктів харчування. У результаті сенсорного аналізу визначають колір, смак, запах, консистенцію харчових продуктів. Дегустаційний метод передбачає апробування харчових продуктів. Результати дегустації залежать від кваліфікації експерта, дотримання умов дегустації: не можна курити, використовувати пахучі речовини, у тому числі парфумерію.
Діючі стандарти передбачають органолептичну оцінку якості продукції порівнянням з еталонами і стандартними зразками.
Значна перевага даного методу – швидкість при отриманні даних, порівняно із використанням хімічного аналізу чи аналізу за допомогою інструментів. Суттєвим недоліком методу – є слабка верифікованість, та значна суб’єктивність[58].
Визначення органолептичних властивостей молока проводили за
ГОСТ 28283-89; готового кисломолочного напою проводили за загальноприйнятою методикою згідно ДСТУ 4343:2004.
Визначення якості рослинної сировини (пелюстки гібіскусу)
Сировину розмістити на білому папері при денному розсіяному або при яскравому штучному освітленні.
Перевірити тип та форму сировини, рівність однорідність, розмір часток, наявність або відсутність пилу, колір. Визначити ступінь однорідності сировини за способом підготовки та кольором.
Описати зовнішній вид сировини. Визначити розміри часток сировини. Лінійкою замірити розміри кількох часток сировини, знайти середнє значення показників.
Проаналізувати запах проб. Для цього сировину розтерти між пальцями. Запах має відповідати виду рослини, не повинен мати затхлого відтінку.
Перевірити наявність домішок.
Визначити смак рослинної сировини. Для аналізу приготувати 10 %-ий відвар[59].
Результати занести до табл.
Таблиця 2.1.
Результати визначення якості рослинної сировини[59]
| Показники якості | Результати аналізу |
| Оганолептичні позники: зовнішній вигляд колір розмір часток запах смак |
Продовження до таблиці 2.1.
| однорідність сировини | |
| Наявність домішок: сміттєвих домішок мінеральних домішок органічних домішок домішок даної сировини домішок інших рослин |
Визначення кислотності молочної сировинититрометричним методом (титрована кислотність). ГОСТ 3624-92
Для визначення кислотності у конічну колбу місткістю 150 або 200 см3 за допомогою піпетки відміряють 10 см3 молока, додають 20 см3 дистильованої води та три краплини 1%-го спиртового розчину фенолфталеїну, ретельно перемішують і титрують розчином гідроксиду натрію (калію) концентрацією 0,1 моль/дм3 до появи слабко-рожевого забарвлення, яке відповідає контрольному еталону і не зникає протягом 1 хв.
Кислотність молока або кисломолочного напою у градусах Тернера визначається об’ємом розчину гідроксиду натрію (калію) концентрацією 0,1 моль/дм3, витраченого на титрування, помноженого на 10.
Розбіжність між двома паралельними вимірюваннями не повинна перевищувати 10Т[60].
Визначення кислотності молочної сировини вимірюванням pH (активна кислотність)
Для визначення рН використовують прилад типу РН-340 та іономір універсальний ЕВ-74.
Проведення аналізу. Перед початком роботи правильність показань приладу перевіряють за буферними розчинами. Близько 40 см3 молочної сировини температурою 20 ± 2 °С поміщають у хімічну склянку, занурюють в неї електроди і через 10... 15 с знімають показання за шкалою приладу.
Для швидкого встановлення показань вимірювання рН молока та іншої молочної сировини проводять за умови безперервного легкого збовтування. Знімання показань за шкалою приладу виконують після повної зупинки стрілки. Після кожного вимірювання електроди датчика промивають дистильованою водою.
За кінцевий результат вимірювання рН приймають середнє арифметичне трьох паралельних вимірювань[60].
Визначення кальцію в молоці комплексонометричним методом (за Дуденковим)
У конічну колбу ємністю 250…300 см3 піпеткою вносять: 5 см3 молока, 90...95 см дистильованої води, 5 см3 8 %-ного розчину гідроксиду натрію. До отриманої суміші з бюретки додають 3,5 см3 0,1 моль/дм3 розчину трилону-Б. Вміст колби перемішують і залишають на 2 хв., після чого на кінчику ножа до неї вносять близько 0,04 г сухої суміші індикатора мурексиду з хлоридом натрію, який забарвлює розчин у бузковий колір. Отриману суміш титрують 0,1 моль/дм3 розчином хлориду кальцію, додаючи його краплинами при безперервному перемішуванні до появи стійкого рожевого забарвлення. Потім проводять зворотне титрування суміші трилоном-Б, додаючи його краплинами при безперервному перемішуванні до появи стійкого бузкового кольору, що не зникає протягом 1 хв. У разі зникнення забарвлення до суміші додають ще одну краплину трилону-Б. Із загального об’єму розчину трилону-Б (3,5 см3+ кількість трилону-Б, що пішов на зворотне титрування) перерахованого на точно 0,1 моль/дм3 розчин, розраховують об’єм 0,1 моль/дм3 розчину хлориду кальцію, використаного на зворотне титрування, і знаходять об’єм трилону-Б, зв’язаного з кальцієм молока.
Опрацювання результатів. Масова частка кальцію, мг %, обчислюється за формулою:
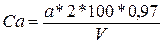 (1)
(1)
де а – об’єм 0,1 моль/дм3 розчину трилону-Б, зв’язаного з кальцієм, см3
2 – маса кальцію, яка відповідає 1см3 0,1 моль/дм3 розчину трилону-Б, мг;
V – об’єм молока, взятого для аналізу, см3[60].
Визначення молочного цукру (лактози) в молоці йодометричним методом (ГОСТ 8764)
Дослідження починають з приготування фільтрату. У мірну колбу ємністю 500 см3 зважують з точністю до 0,01 г 25 г молока (або відміряють 25 см3 молока піпеткою і множать на густину молока), додають до половини колби дистильовану воду і 10 см3 розчину сульфату міді, 4 см3 1 моль/дм3 розчину гідроксиду натрію. Потім суміш перемішують, доводять до позначки водою (при температурі 20 °С), ще раз перемішують і залишають відстоятись протягом 30 хв. Відстояну рідину фільтрують через складчастий паперовий фільтр у суху колбу чи хімічну склянку, виливаючи перші 20...30 см3 фільтрату в каналізацію.
Потім у конічну колбу ємністю 250...300 см3 з притертою або гумовою пробкою піпеткою переносять 50 см3 фільтрату (2,5 г молока). Піпеткою або з бюретки до фільтрату приливають 25 см 0,1 моль/дм3 розчину йоду і повільно, безперервно перемішуючи, до суміші з бюретки вносять 37,5 см3 0,1 моль/дм3 розчину гідроксиду натрію. Колбу закривають пробкою, і залишають у темному місці на 20 хв. При температурі 20 °С. Потім до суміші додають 8 см3 0,5 моль/дм3 розчину соляної кислоти і виділений йод відтитровують 0,1 моль/дм3 розчином тіосульфату натрію. Спочатку темно-коричневу суміш титрують без індикатора до отримання світло-жовтого забарвлення, а потім додають 1 см3 1 %-ного розчину крохмалю і продовжують титрування краплями до моменту, коли від останньої краплі розчину тіосульфату натрію синє забарвлення суміші зникне.
Для проведення холостого досліду в іншу конічну колбу з такою ж ємністю відміряють 25 см3 0,1 моль/дм3 розчину йоду, 25 см води і при безперервному перемішуванні з бюретки додають 37,5 см3 0,1 моль/дм3 розчину гідроксиду натрію. Закривають колбу пробкою і залишають її у темному місці на 20 хв. при температурі 20 °С. Далі визначення проводять так само, як і з фільтратом.
Опрацювання результатів. Масова частка лактози, %:
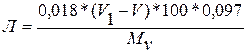 (2)
(2)
де 0,0181 – масова частка лактози, моногідрату, що відповідає 1 см3 0,1 моль/дм3 розчину йоду, г;
V1 ,V – б’єм 0,1 моль/дм3 розчину тіосульфату натрію, що пішов на титрування йоду відповідно в холостому досліді і фільтраті, см3;
0,97 – емпірично встановлена поправка;
Мм – маса молока в 50 см3 фільтрату, яка дорівнює 2,5 г[60].
Визначення масової частки білка у молоці методом формольного титрування
У конічну колбу ємністю 150 або 200 см3 за допомогою піпетки відміряють 20 см3 молока, додають 0,25 см3 2 %-ного спиртового розчину фенолфталеїну, ретельно перемішують і титрують розчином гідроксиду натрію (калію) концентрацією 0,1 моль/дм3до появи слабко-рожевого забарвлення, яке відповідає контрольному еталону. Потім до розчину додають 4 см3 нейтралізованого (свіжо приготованого) 36-40%-ного формаліну і повторно титрують до отримання забарвлення такої ж інтенсивності як і при першому титруванні.
Приготування контрольного еталону здійснюють наступним чином: у конічну колбу ємністю 150 або 200 см3 піпеткою відміряють 20 см3 молока, до нього додають 0,5 см3 2,5 %-ного розчину сірчанокислого кобальту і ретельно перемішують. Еталон придатний для роботи протягом одного заняття.
Масова частка білка у молоці в процентах визначається як добуток емпіричного коефіцієнта 0,959 та об’єму розчину гідроксиду натрію або калію концентрацією 0,1 моль/дм3, що пішов на титрування в присутності формаліну[60].
Методика отримання екстракту з гібіскусу
1. Визначення оптимальної температури екстрагування.
Досліджувані температури: 20 0С, 40 0С, 60 0С, 80 0С.
Зважуємо 10 г гібіскусу (4 порції). Поміщаємо кожну пробу у стаканчик і доливаємо дистильованої води у співвідношенні 1:10. Процес екстрагування проводити протягом 30 хв при зазначеній температурі. Вміст кожного стаканчика фільтруємо і в отриманих фільтратах визначаємо такі показники: вміст сухих речовин по рефрактометру, рН по рН-метру і оптичну густину. Отримані показники заносимо у таблицю
Таблиця 2.2.
Основні показники отриманих екстрактів з гібіскусу залежно від температури процесу
| Показники | Температура процесу, С | |||
| рН | ||||
| СР, % | ||||
| Оптична густина, од.опт.густ |
Оптимальну температуру екстрагування визначаємо за найбільшим вмістом сухих речовин у екстракті.
2. Визначення оптимальної тривалості екстрагування.
Процес проводимо при оптимальній температурі й таких тривалостях: 30 хв, 60 хв, 90 хв, 120 хв. Зважуємо такох 10 г гібіскусу (4 порції), гідромодуль 1:10. Після зазначеної тривалості процесу екстрагування вміст стаканчиків фільтруємо і у фільтратах визначаємо такі ж показники як і у 1 випадку. Отримані дані заносимо до таблиці:
Таблиця 2.3.
Основні показники отриманих екстрактів залежно від тривалості процесу
| Показники | Тривалість процесу, хв | |||
| рН | ||||
| СР, % | ||||
| Оптична густина, од.опт.густ |
Оптимальний час екстрагування визначаємо за найбільшим вмістом сухих речовин у екстракті.
3. Визначення залежності показників екстрагування від виду екстрагента.
При оптимальній температурі і оптимальній тривалості процесу встановлюємо залежність показників від виду екстрагенту: дистильована вода, 10 %-ий водно-спиртовий розчин, 20 %-ий водно-спиртовий розчин, 40 %-ий водно-спиртовий розчин. Зважують 10 г гібіскусу (4 порції) поміщають у 4 стаканчики і додають екстрагент:
в перший – 100 мл води;
в другий – 100 мл 10 %-го розчину;
в третій – 100 мл 20 %-го розчину;
в четвертий – 100 мл 40 %-го розчину.
При оптимальній температурі і оптимальній тривалості процесу проводять екстрагування з чотирма екстрагентами. Після закінчення екстрагування вміст стаканчиків фільтруємо і у фільтратах визначаємо такі ж показники, які в попередніх двох випадка і заносимо результати до таблиці:
Таблиця 2.4
Залежність основних показників від виду екстрагента
| Показники | Вид екстрагенту | |||
| вода | 10 %-ий водно-спиртовий р-н | 20 %-ий водно-спиртовий р-н | 40 %-ий водно-спиртовий р-н | |
| рН | ||||
| СР, % | ||||
| Оптична густина, од.опт.густ |
За даними останньої таблиці визначаємо найбільш ефективний екстрагент за результатами максимального вмісту сухих речивин.
Висновок: на піставі аналізу даних усіх трьох таблиць пропонуємо оптимальні значення основних параметрів екстрагування:
температура процесу – ….
тривалість процесу – ….
вид екстрагенту – ….
Рефрактометричний метод визначення сухих речовин
Метод базований на визначенні масової частки сухих речовин випробувальної рідини за шкалою рефрактометра за температури 20°С після проведення в пробі продукції повної інверсії.
На нижню призму рефрактометра наносять скляною паличкою 2 або 3 краплі випробувальної рідини. Верхню частину призми опускають, щільно прикладають до нижньої нерухомої частини призми і проводять відлік за шкалою рефрактометра. Під час відліку показників приладу фіксують температуру, за якої проводять випробування.
Визначення загального вмісту фенольних речовин у соках (екстрактах), фруктах і плодах
У мірну колбу місткістю 100 см3 поміщають 1 см3 світлого соку або 1 см3 темнозабарвленого соку, попередньо розведеного в 5 разів ( 2 см3 соку і
8 см3 дистильованої води розводять у пробірці). Потім у мірну колбу додають 2 см3 реактиву Фоліна-Чокальтеу і 10 см3 20 % розчину соди. Об’єм доводять водою до мітки, перемішують і витримують 30 хвилин. Після витримки в розчині визначають оптичну густину розчину при довжині хвилі 630 нм і довжині кювети 10 мм на фотоелектокалориметрі або спектрофотометрі. При значеннях оптичної густини, більших 0,5 одиниць, аналізований зразок слід додатково розбавити, враховуючи це при розрахунку вмісту фенольних речовин. Як розчин порівняння використовують реактив, приготований з
1 см3 реактиву Фоліна-Чокальтеу і 10 см3 20 % розчину соди, доведених в мірній колбі до 100 см3.
При аналізі плодово-ягідної сировини спочатку готують витяжку. На технічних вагах зважують 10 г сировини, розтирають у порцеляновій ступці протягом 10 хвилин, поступово додають при розтиранні 10 см3 води. Подрібнену наважку переносять кількісно в мірну колбу місткість 100 см3, доводять дистильованою водою до мітки, перемішують і фільтрують через паперовий складчастий фільтр. На аналіз відміряють 1 см3 в мірну колбу місткістю 100 см3 і додають реактиви, як зазначено вище. При визначенні загального вмісту фенольних речовин в темнозабарвленій сировині, наприклад у чорній сородині, приготований фільтрат необхідно розбавити в 5 разів, як сік при цьому розбавлення слід враховувати в розрахунку фенольних речовин.
Вміст фенольних речовин визначають за калібрувальним графіком. Для пробудови калібрувального графіка відмірюють по 1, 2, 5, 10, 20 см3 стандартного розчину енотаніна в мірні колби на 100 см3, що відповідає 0,3; 0,6; 1,5; 3,0; 6,0 мг/дм3 таніну. У кожну колбу додають 1 см3 реактиву Фоліна-Чекальтеу, 10 см3 20 % розчину соди, вміст колб доводять до мітки, перемішують. Черех 30 хвилин витримки визначають оптичну густину розчинів при тих же параметрах. Використовуючи отримані результати, будують калібрувальну криву, відкладаючи на осі абсцис вміст таніну в досліджуваних зразках, а на осі ординат – значення оптичної густини.
За відсутності енотаніну калібрувальний графік, з певною похибкою, що вноситься використовуваним приладом, може бути побудований за наступними даними:
вісь абсцис – 0,3; 0,6; 1,5; 3,0; 6,0 мг/дм3 таніну;
вісь ординат – 0,024; 0,046; 0,108; 0,214; 0,424 – одиниць оптичної густини.
Для розрахунку вмісту фенольних речовин необхідно концентрацію таніну, знайдену за калібрувальним графіком, помножити на коефіцієнт розбавлення: для темнозабарвлених соків – 500, а для світло забарвлених соків – 100. Для плодово-ягідної сировини коефіцієнт розбавлення 10 в перерахунку на 1 г сировини і 1000 в перерахунку на 100 г сировини[61].
Визначення вмісту антоціанів у рослинній сировині та екстрактах методом диференційної спектроскопії прри двох значеннях рН
Буферний розчин № 1( рН 1,0): 0,405 мг КСІ. 1,238 мл НСІ (конц.), довести водою до 100 мл.
Буферний розчин № 2 (рН 4,5): 1,64 г натрію ацетату в 100 мл води і НСІ до рН 4,5,;
Послідовність виконання роботи.
1. Наважку 1,00 г сировини подрібнити до розміру часток, що проходять через сито з отворами діаметром 2 мм, помістити у колбу зі шліфом місткістю 200 мл, додати 50 мл 70% етилового спирту. Колбу приєднати до зворотного холодильника і нагрівати на водяній бані протягам 60 хв, періодично збовтуючи.
2. Гарячі витяги профільтрувати у мірну колбу місткістю 50 мл. Після охолодження довести водою до мітки.
3. До буферних розчинів № 1 або № 2 додати екстракт, визначити оптичну густину при 510 нм і 700 нм з відповідним буфером в якості розчину порівняння на спектрофотометрі. Для кожного зразка визначення повторити тричі.
5. Сумарний вміст антоціанових пігментів розрахувати за формулою:
А= (А510 pH 1,0 – А700 pH 1,0) – (А510 pH 4,5 – А700 pH 4,5)
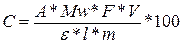 , (3)
, (3)
де С – масова частка антоціанів, 5 %; А – поглинання; 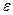 і Mw – коефіцієнт молярного поглинання і молекулярна маса антоціаніна, що використано в якості стандарту (для ціанідин-3-глюкозиду – 26900 і 449,2 відповідно); F – розведення; V – об’єм витягу, мл; l – довжина кювети, см;
і Mw – коефіцієнт молярного поглинання і молекулярна маса антоціаніна, що використано в якості стандарту (для ціанідин-3-глюкозиду – 26900 і 449,2 відповідно); F – розведення; V – об’єм витягу, мл; l – довжина кювети, см;
m – маса зразка, мг[59].
Кількісний аналіз на вміст пектинових речовин у рослинній сировині
На технічних терезах зважують наважку масою 25 г вологого або10 г сухого досліджуваного матеріалу, розтирають в ступці до однорідного стану і переносять в колбу місткістю 150 см3. Додають у колбу 100 см3 дистильованої води при температурі 40 °С і витримують на водяній бані 30 хвилин, фільтрують і знову заливають 75 см3 води, а потім ще раз 50…60 см3 води. Отримані екстракти збирають у мірну колбу на 250 см3 і доводять до мітки дистильованою водою. Отриманий розчин гідратованих пектинів використовують для подальших аналізів.
Для визначення масової частки у рослинній сировині протопектину і пектової кислоти залишок на фільтрі заливають 50 см3 розчину НС1 (0,3 моль/дм3) і нагрівають із зворотним холодильником на протязі 30 хвилин на
киплячій водяній бані у колбі місткістю 250 см3.
Потім екстракт фільтрують через складчастий фільтр і переносять у мірну колбу на 500 см3. Залишок на фільтрі 3-4 рази промивають водою (наближено 75 см3) промивні води фільтрують в ту ж саму колбу.
Фільтр із залишком рослинного матеріалу переносять в конічну колбу,
заливають 50 см3 0,06 моль/дм3 розчину лимоннокислого амонію і знову розміщують на 30 хвилин на киплячу водяну баню. По закінчені часу гідролізатфільтрують в ту ж мірну колбу, що і в перший раз. Гідролізати в усіх випадках фільтруються без попереднього охолодження.
Далі фільтрат нейтралізують 10 %-ним розчином NaOH (в середньому 5 см3) і вміст колби доводять до мітки дистильованою водою. Пектинові речовини, включаючи протопектин, знаходяться у формі пектинової кислоти. Потім з мірної колби беруть 50 см3 фільтрату, додають 50 см3 0,1 моль/дм3 розчину натрій гідроксиду (NaOH) і залишають на ніч при кімнатній температурі для омилення метоксильних груп. На наступний день розчин нейтралізують 50 см3 оцтової кислоти (1 моль/дм3) і додають 50 см3 СаС12 (2 моль/дм3). Для повноти перебігу реакції з СаС12 пектинової кислоти розчин відразу кип’ятять 5 хвилин або залишають на 1 годину. Після кип’ятіння утворився осад пектату кальцію фільтрують через висушений до постійної маси беззольногої фільтру.
Обчислення результатів. Осад на фільтрі промивають киплячою водою до зникнення позитивної реакції на хлор. Потім осад пектату кальцію разом з фільтром переносять у бюкс і при температурі 100 °С і доводять до постійної маси. Виходячи з маси кальцій пектату, розраховують вміст пектину (Xn),% за формулою:
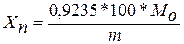 , (4)
, (4)
де Мо – маса осаду пектату кальцію, г; m – маса наважки, г; 0,9235 – коефіцієнт, що враховує масу кальцію в молекулі пектату кальцію[62].
Визначення вмісту вітаміну С у забарвлених витягах
Аскорбінову кислоту забарвлених витягів титрують розчином 2,6-дихлорфеноліндофенола (фарби) за присутності хлороформу, дихлоретану чи толуолу.
Наважку сировини (10 г) екстрагують як зазвичай і доводять об’єм кстракту до 100-200 см3 розчином щавлевої кислоти. Після фільтрування відбирають піпеткою 5 см3 забарвленого екстракту в пробірки (діаметр 20-25 мм, довжина 150 мм) і туди ж додають по 5 см3 хімічно чистого хлороформу. Потім витяжку титрують розчином фарби, обережно перемішуючи, нахиляючи пробірку, прикриту пробкою.
При появі першого рожевого забарвлення у шарі хлороформу титрування вважають закінченим. Одночасно проводять таке ж титрування контрольних проб з тією різницею, що замість екстракту в пробірки додають суміш розчинів соляної і щавлевої кислот у співвідношенні об'ємів 1:5.
Кількість аскорбінової кислоти (Х) у зразку осмислюють за формулою:
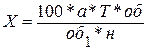 , (5)
, (5)
де: а – об’єм фарби ( з вирахуванням поправки на титрування чистого розчинника), який пішов на титрування екстракту, см3; Т – титр фарби за аскорбіновою кислотою; об – загальний об’єм витягу, см3; об1 – об’єм екстракту, взятого на титрування, см3; н – наважка матеріалу, г[63].
Дата добавления: 2015-06-05; просмотров: 2509;