Взаимопревращение функциональных групп как стратегический метод в полном синтезе.
В начальный период развития органического синтеза было естественно выстраивать синтетическую цепочку, используя в качестве исходного соединения то или иное вещество, выделяемое из природных источников. Именно на этой основе в 1911 г. Вильштеттером [21а] был осуществлен синтез цикло-октатетраена (137). Исходным соединением для этого синтеза послужил алкалоид псевдопельтьерин (138), выделенный из корней гранатового дерева. Это соединение уже содержало 8-членный цикл, и поэтому представлялось наиболее естественным предшественником для получения 137. Задача Виль-цггеттера состояла в преобразование функциональности, имеющейся в алкалоиде 138, в систему четырех двойных связей целевого продукта 137. Первая двойная связь была создана в результате восстановления кетогруппы с последующей дегидратацией, а построение остальных двойных связей было осуществлено с помощью последовательности таких простых реакций, как исчерпывающее метилирование, элиминирование по Гофману, бромирова-ние и т. д., как это показано на схеме 2.65. Все стадии этого 10-стадийного синтеза суть трансформации функциональных групп, две из которых неизо-гипсические (восстановление карбонила и присоединение брома), а остальные — изогипсические.
Но надо сказать, что подобного рода частичные синтезы, основанные на преобразовании уже почти готовой заготовки в целевое соединение путем пере-функционализации, — большая редкость в истории органического синтеза, по крайней мере, применительно к карбоциклическим системам, поскольку лишь в очень редких случаях удавалось найти столь прямолинейное соответствие между структурами целевой молекулы и доступного природного соединения.
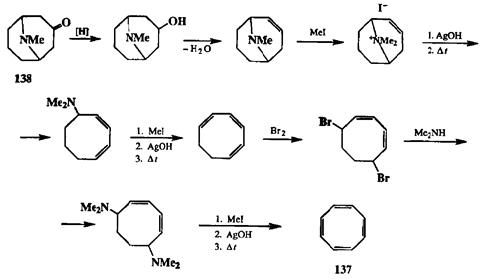 Схема 2.65
Схема 2.65
|
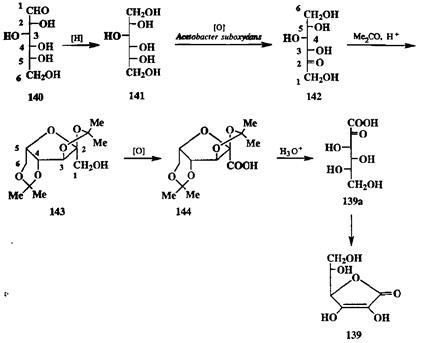 Схема 2.66
Схема 2.66
|
Примером большей области, почти целиком построеной на частичных синтезах, может служить синтетическая химия углеводов. В этих исследованиях приходится решать задачи двух типов, а именно: синтез природных моносахарвдов и их аналогов и сборку олигомерных (олигосахариды) и полимерных (полисахариды) систем из моносахаридных мономеров. Природные моносахариды весьма разнообразны по структуре, но различия между ними сводятся почти исключительно к различиям в природе и расположении функциональных групп, а также различиям в конфигурации асимметрических центров. В то же время большинство этих моносахаридов имеет одинаковый или очень сходный углеродный скелет — цепь из пяти или шести углеродных атомов. Многие природные моносахариды легкодоступны (как, например, D-глюкоза, L-apaбиноза и ряд других). Для их превращения во множество других моносахаридов обычно достаточно изменить характер нескольких функциональных групп (скажем, заменить спиртовую группу на аминогруппу или окислить первичную спиртовую группу до карбоксильной) и изменить конфигурацию одного или нескольких асимметрических центров. Поэтому нет никакого резона заново создавать углеродный скелет, размещать на нем многочисленные функциональные группы и обеспечивать необходимые конфигурации асимметрических центров, если большинство из этих задач уже решены Природой при биосинтезе, скажем, D-глюкозы. Поэтому генеральный путь синтеза моносахаридов обычно предполагает серию последовательных превращений, направленных на трансформацию функциональных групп и изменение конфигурации асимметрических центров в молекулах исходных природных моносахаридов.
Рассмотрим для иллюстрации промышленный синтез аскорбиновой кислоты (139) из D-глюкозы (140) (схема 2.66). Каталитическое гидрирование 140 приводит к шестиатомному спирту — D-сорбиту (141). Сорбит подвергают далее микробиологическому окислению, в результате которого селективно вводится кетогруппа в положение 2. Образовавшийся изомер глюкозы, L-сорбозу (142), превращают в днизопролилиденовос производное 143, защищая тем самым все функциональные группы, кроме одной, первичной спиртовой при С-1 (бывшего С-6 исходной глюкозы). Эту группу далее окисляют до карбоксильной, что дает защищенное производное 144. Удаление защитных групп кислотным гидролизом приводит к аскорбиновой кислоте (139а), которая самопроизвольно превращается в енольную форму лактона 139.
Как видно, в этом синтезе основные стадии представляют собой неизо-гнпсические трансформации функциональных групп — это одна реакция восстановления (получение 141) и две реакции окисления (получение 142 и 144). Изогипсические реакции постановки и снятия изопропилиденовой защиты, а также выбор столь необычного окисляющего агента, как Acetobacter suboxidans, предназначены для обеспечения селективности указанных трансформирующих реакций полифункционального субстрата.
Олиго- и полихариды построены из остатков моносахаридов, соединенных через атом кислорода гликозидной связью. Поэтому ключевым моментом синтеза таких систем является создание гликозидной связи между моносахаридными звеньями. Формальная схема сборки такой связи может быть проиллюстрирована образованием молекулы дисахарида лактозы (145,молочный сахар) из моносахаридных предшественников — D-галактозы (146)и D-глюкозы (140)(схема 2.67).
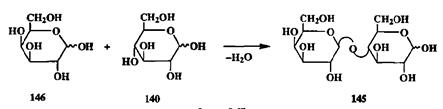 Схема 2.67
Схема 2.67
|
Как видим, здесь также не требуется создания новой связи С-С, и задача состоит в трансформации функциональных групп: превращении голуаце-тального гидроксила галактозы в гликозид и спиртового гидроксила при С-4 в производное с галактозильным остовом. В общей органической химии это самые тривиальные превращения. Но это далеко не тривиальная задача, когда речь идет о гликозидной связи. Сгереоселективное образование такой связи представляет собой одну из центральных проблем химии углеводов, и многие сотни работ посвящены разработке методов эффективного решения этой проблемы (см., например, обсуждение в монографиях [21Ь]).
Аналогична ситуация в химии пептидов и белков: здесь также синтез строится на сборке межмономерной (пептидной) связи между аминокислотами, используемыми в качестве доступных строительных блоков. Точно также и в химии третьего важнейшего класса биополимеров — нуклеиновых кислот — центральным вопросом синтеза является построение межмономерной фос-фодиэфирной связи, т. е. в чисто органохимических терминах «всего-навсего» трансформация спиртовых гидроксилов в эфиры фосфорной кислоты.
Из всего сказанного должно быть ясно, что трансформация функциональных групп может играть решающую, а отнюдь не подсобную роль во многих синтезах. В определенных (и весьма немаловажных!) областях химии природных соединений подобного рода превращение может представлять собой самостоятельную проблему принципиального значения, решение которой требует наличия разнообразных и подчас очень изощренных методических разработок.
Дата добавления: 2015-04-05; просмотров: 2033;