Неизогипсические трансформации как пути переходов между различными уровнями окисления
В этой группе наиболее значимыми для синтеза являются такие превращения кислородсодержащих соединений, как окисление спиртов до карбонильных соединений или карбоновых кислот и обратные им превращения. Поэтому неудивительно то особое внимание, которое синтетики уделяли и уделяют разработке безотказных методов реализации таких переходов. Эти усилия не оказались напрасными — правомерно утверждать, что задача эффективного и селективного проведения любого из упомянутых превращений может быть успешно решена при практически любой комбинации осложняющих факторов (лабильность субстратата или продукта реакции, наличие других реагирующих групп, стерическис препятствия).
Особо интенсивно разрабатывались новые методы в применении к задаче окисления спиртов. В цитированной выше монографии Ларока [19е] перечислено более 140 процедур дтя окисления спиртов с использованием самых различных окислительных систем. В этом перечне можно найти также несколько десятков методик, специально предназначенных для окисления определенных типов спиртов, таких, как первичные или вторичные, аллиловые или гомоаллиловые.
Чаще всего окисление спиртов проводят с помощью хромового ангидрида и его комплексов [19Е]- Существует также целый набор удобных методов окисления спиртов, основанный на использовании диметилсульфоксида в качестве окислителя в присутствии различных кислот Льюиса — это реакция Принтера— Моффата (схема 2.60) [19dJ. В этой реакции ключевой стадией является образование алкоксисульфолийилида в качестве интермедиата, который далее распадается, давая карбонильное производное и диметил-сульфид. Диметилсульфоксид в присутствии оснований способен также окислять в карбонильные соединения и другие производные первого уровня окисления, такие, как алкилгалогениды и алкилтозилаты, через стадию образования того же интермедиата, но эта реакция эффективно протекает лишь с первичными субстратами [19g].
Диоксид марганца МпО2 является специфическим реагентом для окисления аллиловых спиртов до альдегидов (19Л. При этом полностью исключается возможность переокисления до карбоновых кислот, что не всегда легко Достижимо с помощью других окислителей. Примечательно, однако, что та же реакция, если проводить ее присутствии цианид-иона, применяется как удобный метод окисления аллиловых спиртов до карбоновых кислот или их метиловых эфиров [I9h] (в последнем случае метанол используется в качестве растворителя). В этом варианте проведения окисления первоначально также образуется альдегид, но в присутствии цианид-иона он дает циангид-рин, который содержит группировку аллилового спирта и поэтому способен окисляться далее под действием того же окислителя (см. схему 2.60).
Что же касается общих методов окисления альдегидов в карбоновые кислоты, то это превращение можно проводить с помощью самых различных окислителей, например О2, Ag2O, KMnO4, NaC102 и т. д.
Обратное превращение карбонильных производных в спирты обычно проводится с помощью комплексных гидридов металлов, таких, как LiAlU,, NaBH4 и их многочисленные производные. Активность и селективность подобных доноров гидрид-иона можно изменять в широких пределах благодаря возможности вариаций конкретной природы этих реагентов, что имеет Огромное значение для управления селективностью реакции восстановления фб этом см. разд. 2.4.1).
Надежность методов осуществления перечисленных трансформаций по-зйоляет считать кислородсодержащие функции разных уровней окисления Синтетически эквивалентными. Иными словами, если, например, в целевой молекуле в некотором месте должна находиться кетогруппа, то адекватным решением задачи построения этого фрагмента может служить синтез соответствующего вторичного спирта (и наоборот), а с учетом широких возможностей изогипсических трансформаций первого уровня окисления — почти любого функционального производного этого уровня окисления. В синтезе азотсодержащих производных очень важны неизогипсические трансформации азотсодержщих функций различных степеней окисления. Так, один из общих методов получения аминов основан на восстановлении азотсодержащих производных кислот (нитрилы, амиды) или альдегидов и кетонов (имины).
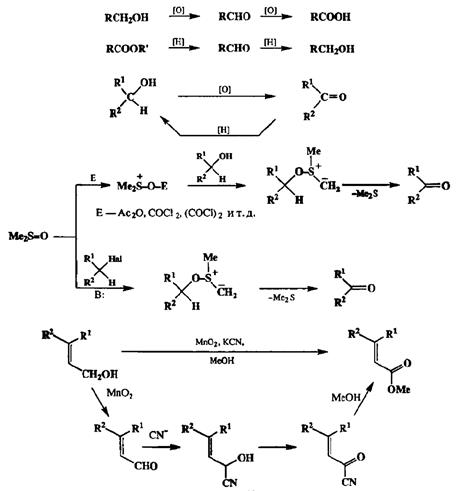 Схема 2.60
Схема 2.60
|
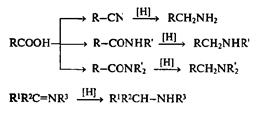
Заметим попутно, что синтез аминов может выполняться и с помощью Другой серии реакций, где изогипсическому превращению подвергается азотный фрагамент, а состояние окисления связанного с ним атома углерода не изменяется.
Как уже отмечалось ранее, связь С-С легко «встраивается» в собираемую молекулу с помощью одного из набора стандартных методов, таких, например, как алкилирование ацетиленидов или этинилирование карбонильных соединений. Благодаря легкости осуществления селективного превращения тройной связи в двойную (каталитическое гидрирование или восстановление металлами) правомерно рассматривать алкины как синтетические эквиваленты алкенов. В свою очередь, алкены могут быть легко восстановлены в алканы. Итак, алкены и алкины могут считаться синтетическими эквивалентами алканов, и все методы образования углерод-углеродной связи, используемые в синтезе алкенов и алкинов, в равной степени пригодны и для получения соответствующих насыщенных производных.
Что касается окислительных трансформаций алкенов, то здесь особое место занимает их превращение в эпоксиды. Для этой цели в промышленности используют каталитическое окисление кислородом, а в лаборатории — над-кислотами, среди которых особенно эффективна .м-хлорнадбснзойная кислота. Эпоксиды могут легко превращаться в 1,2-гликоли в условиях кислотного гидролиза. Поскольку эта реакция протекает с обращением конфигурации по одному из атомов углерода, то суммарный результат превращения ал-кен → эпоксид → 1,2-гликоль соответствует анти-присоединению гидро-ксильных групп. Можно также осуществить и син-присоединение, для чего требуется использовать в качестве окислителей реагенты типа КМпО4 или OsO4. Различие в сгерическом результате этих двух формально сходных процессов вполне объяснимо в рамках представлений о механизмах этих реакций, показанных на схеме 2.61.
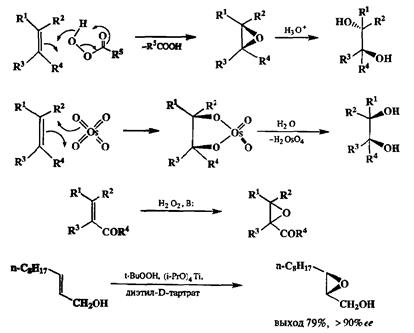 Схема 2.61
Схема 2.61
|
Разнообразие разработанных методов окисления алкенов (в монографии Ларока [19i] перечислено более 80 различных окислительных систем) делает возможным проведение селективного окисления олефинового фрагмента при наличии в структуре самых различных функциональных групп. Так, использование пероксида водорода в щелочных условиях позволяет селектив-tio эпоксидировать двойную связь в се,р-непрсдельных карбонильных соединениях.
Особенно большое внимание было уделено разработке путей энантиосе-йективного эпоксидирования. На этом направлении наиболее успешными явились разработки группы Шарплесса, в результате которых была найдено, что исключительно высокая энантиоселективность окисления аллиловых спиртов может быть достигнута, если использовать в качестве окислителя систему /ярет-бутилгидропероксид/тетраизопропилат титана и проводить реакцию в присутствии D- или L-диэтилтартрата (схема 2.61). В этих условиях реакция проходит с хорошим химическим выходом и дает соответствующие эпоксиды высокой оптической чистоты (> 90% ее) [19j].
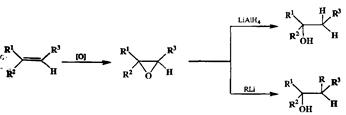 Схема 2.62
Схема 2.62
|
Превращение алкенов в эпоксиды важно не только как один из самых надежных способов осуществления перехода с первого на второй уровень окис-дения. Хорошо известно, что раскрытие эпокевдов под действием нуклеофи-лов протекает как атака нуклеофила по менее замещенному атому углерода с обращением конфигурации по этому центру. Это дает возможность, в частности, применять последовательность превращений эпоксидирование двойной связи/гидридное восстановление эпоксида как метод направленного синтеза спиртов (схема 2.62). Этот способ особенно важен для синтеза энантиомерно чистых спиртов из эпоксидов, получаемых окислением по Шарплессу.
На схеме 2.62 показано еще одно превращение эпоксидного фрагмента, а именно раскрытие оксиранового цикла под действием карбанионных реагентов. В этом превращении эпоксидная группировка выступает в роли эквивалента р-алкоксикарбокатиона, и реакции, основанные на использовании свойств эпоксидов как элсктрофилов такого структурного типа, очень Йкроко применяются в синтетической практике. Легко заметить, что обе Стадии превращений, показанные на схеме 2.62, относятся к разряду неизо-гипсических реакций, причем результатом первой из них является превращение исходного нуклеофильного реагента (алкена) в электрофильный (эпоксид) за счет окислительной трансформации, а на второй стадии — восстановительной — осуществляется возврат на исходный уровень окисления с образованием спиртов нужного строения.
Множество других неизогипсических превращений алкенов основано на использовании электрофильного присоединения. Среди них стоит отметить присоединение галогенов, одну из самых старых реакций непредельных соединений. Получаемые при этом 1,2-дигалогениды являются соединениями второго уровня окисления, и они широко применяются как ключевые полупродукты в синтезе винилгалогенидов (в частности, винилхлорида — мономера для получения ПВХ) и ацетиленов:
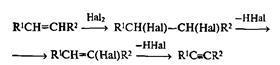
Мы уже неоднократно могли убедиться в том, что в результате большинства реакций образования связи С-С получаются продукты, которые содержат какие-либо функцион&чьные группы (к числу немногих исключений принадлежит синтез Вюрца и алкилирование по Фриделю—Крафтсу). Поэтому необходимой составной частью набора инструментов синтетика являются методы исчерпывающего восстановления, результатом которых является удаление функции (переход на нулевой уровень окисления) после того, как она сыграла свою роль. К числу таких методов относится, в частности, уже упоминавшееся гидрирование алкенов и алкинов. Назовем еще некоторые из практически важных маршрутов превращения той или иной функции в алкановый фрагмент.
Прямое восстановительное удаление гидроксильной функции легко осуществимо лишь для третичных или бензильных спиртов, обладающих повышенной способностью к образованию в кислой среде карбокатионных интермедиатов. Последние способны реагировагь с такими активными донорами гидрид-иона, как, например, триэтилсилан, с образованием соответствующего углеводорода. Эта процедура, известная под названии «ионное гидрирование», довольно широко применяется в синтетической практике [19k]:
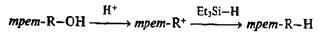
Для восстановления первичных или вторичных спиртов их необходимо сначала превратить в соответствующие алкилгалогениды или алкилсульфо-наты, которые далее способны легко претерпевать гидрогенолиз под действием комплексных гидридов, например алюмогидрида лития [191]:
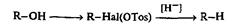
Для восстановления первичных аллиловых спиртов особенно удобна методика, включающая обработку исходного субстрата комплексом пиридин-SO3 с последующим восстановлением промежуточно образующегося суль-фоэфира алюмогидридом лития:
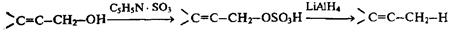
Тиопроизводные, такие, как меркаптаны, сульфиды, тиоацетали, очень легко подвергаются гидрогенолизу под действием никеля Ренея. Для восстановительного удаления сульфонильной группы особенно эффективно восстановление металлами в жидком аммиаке:
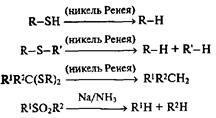
Хорошо известно, что различные серосодержащие производные благодаря способности атома серы стабилизировать как карбанионный, так и карбокатионный центры на соседнем атоме углерода, являются ключевыми реагентами во многих методах образования связи С-С. Именно этим и обусловлена высокая синтетическая значимость показанных выше трансформаций тиопроизводных.
Прямой переход со второго уровня окисления на нулевой уровень легче всего осуществим для карбонильных производных либо через стадии образования тиоацеталей и их гидрогенолиза (см. выше), либо по классической реакции Кижнера—Вольфа путем обработки гидразином в присутствии сильного основания. Препаративно важной является также возможность превращения альдегидов и кетонов в алкены путем восстановительного расщепления тозилгидразонов под действием метиллития (реакция Шапиро) [19т]. Какпоказано на схеме 2.63, непосредственным результатом реакции является образование виниллитиевых производных и именно для генерации последних чаще всего и используется этот метод.
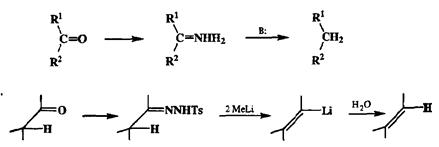 Схема 2.63
Схема 2.63
|
Пожалуй, можно не продолжать перечня возможностей взаимопревращений функциональных групп — приниципиальная сторона использования этих переходов в синтезе уже должна быть ясна. Отметим только, что несмотря на обилие существующих методов трансформаций, интенсивность исследований в этой области не снижается. При этом целью является далеко не только отработка чисто методических вопросов (повышение эффективности и общности реакции, решение задач ее селективного проведения, нахождение более дешевых реагентов и т. п.). Очень большое внимание уделяется также поиску путей реализции новых, иногда совсем нетривиальных переходов от функции к функции. В результате таких работ в арсенал синтетика включаются трансформации, резко сокращающие маршрут перехода из ячейки в ячейку данного «этажа» (уровня окисления) или открывающие новые переходы («лифты») с «этажа на этаж».
Неплохой иллюстрацией полезности поисков в этих направлениях могут служить почти произвольно выбранные примеры реакций (1)—(6):
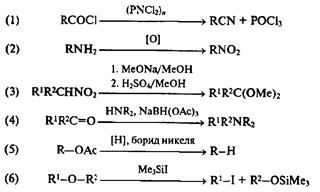
Синтетическое значение реакции (1) [20a] заключается в том, что она сокращает на одну стадию превращение ацилхлоридов в соответствующие нитрилы — превращение, которое традиционно проводится по не очень удобной двустадийной схеме (получение амида и его дегидратация).
Реакция (2) [20Ь] — это пример нового превращения, ранее неосуществимого в ряду алифатических аминов. Трансформация нитрогруппы в карбонильную — это хорошо известная реакция Нефа. Вариант ее, представленный реакцией (3) [20с],дает возможность сразу получать защищенную карбонильную группу в результате этого превращения. Учет возможности проведения последовательности реакций (2) + (3) позволяет рассматривать фрагмент CHNH2 как эквивалент защищенной карбонильной группы.
Трансформация (4) — восстановительное аминирование, это ничто иное, как последовательность стадий получения имина in situ и его восстановления в амин [20d]. Очевидны преимущества проведения этих двух стадий как ре-акцийв одном реакционном сосуде.
Обнаружение способности борида никеля (легко получаемого in situ восстановлением хлорида никеля боргидридом натрия) служить эффективным катализатором гидрогенолиза ацетатов аллильных или третичных спиртов [(реакция (5)] [20е], существенно упрощает применение этого пути перехода спервого уровня окисления на нулевой по сравнению с вышеописанными Способами проведения этого превращения.
Реакция (6) [20f] — это всего лишь еще один пример расщепления простого эфира под действием кислот Льюиса. Однако в отличие от других кислот Льюиса,требующих довольно жестких условий проведения реакции, триме-тилсилилиодид вызывает это превращение уже при комнатной температуре. Благодаря этому обстоятельству, которое, вообще говоря, могло бы показаться второстепенным, мегоксигруппа, ранее считавшейся тупиковой для осуществления трансформационных переходов, теперь может рассматриваться как один из удобных вариантов временной защиты спиртовой функции.
Итак, стремление связать отношениями эквивалентности как можно большее разнообразие функциональных групп — одна из движущих сил в разработке все новых методов трансформационных превращений. Конечная цель таких разработок — создание банка стандартных процедур, позволяющих в 1-2 операции взаимопревращать любые функции. Надо отметить, что, хотя ситуация в этой области еще далека от такого идеала, уже сейчас для большинства функций решение задач трансформационных переходов хорошо обеспечено достаточно богатым арсеналом методов. Это позволяет при планировании синтезов уверенно пользоваться следующими простыми правилами:
1. Любые функциональные группы, особенно относящиеся к одному уровню окисления, можно считать синтетически эквивалентными, так что правомерны их ретросинтетические превращения. Это означает, что если, например, в целевой молекуле имеется гидроксильная группа, то при ретросинтетиче-ском анализе допустимо ее трансформировать, скажем, в галогенид, карбонильную группу, двойную связь или эпоксид и т. д. Все получаемые при таких операциях субструктуры могут считаться эквивалентными, так что синтез любой из них уже будет являться решением проблемы синтеза целевой молекулы. Ясно, что такой подход резко расширяет возможности выбора конструктивных реакций, ведущих к построению рассматриваемого фрагмента структуры.
2. Поскольку любая функция может быть удалена, то в любое положение насыщенного фрагмента целевой молекулы может быть ретросинтетически «вставлена» функция, обеспечивающая возможность применения того или иного метода создания связи С—С в избранном фрагменте.
Вдумчивый читатель мог заметить, что в то время как существует множество способов восстановительных трансформаций различных функций с образованием насыщенного углеводородного фрагмента, мы ни разу не упомянули о возможности обратной трансформации — перехода с уровня 0 к Производным более высокого уровня окисления. Подобное умолчание не случайно. Дело в том, что введение функции в заданное положение алканово-го фрагмента (синтетически, а не умозрительно, в ходе ретросинтетического анализа) в настоящее время весьма затруднительно. Природа затруднений не в отсутствии способов проведения замещения по насыщенному атому углерода — такие реакции есть: это классическое хлорирование, нитрование или окисление. Проблема в другом: связей С-Н, способных вступать в такие реакции в любом углеводороде, множество, так что затруднение связано с невозможностью в общем случае обеспечить селективную функционашзацию лишь одного алканового фрагмента из числа имеющихся в молекуле*.
Напротив, при наличии какой-либо функции сравнительно нетрудно подобрать реакцию, позволяющую ввести дополнительную функцию по соседству с первой. Примером подобного рода нсизогапсических трансформаций может служить аллильное окисление алкенов диоксидом селена до соответствующих а,р-непредельных альдегидов, а-бромирование кетонов или карбоновых кислот (реакция Геля—Фольгарта—Зелинского), а также упоминавшееся выше (схема 2.56) превращение алкенов в аллилбромиды под действием N-бромсукцинимида (NBS) (схема 2.64).
Здесь уместно добавить, что за последние десятилетия наметился значительный прогресс и в разработке подходов, позволяющих обеспечить селективную функционализацию по насыщенному фрагменту, не содержащему активирующих групп. Эго удастся сделать благодаря дизайну специфических структур субстратов, в которых требуемое превращения протекает как внутримолекулярный процесс, селективность которого обеспечивается сближением реагирующих центров. Подробнее этот подход рассмотрен в гл. 4.
До сих пор мы рассматривали методы трансформации функциональных групп как некоторые подсобные инструменты, так или иначе сопряженные с методами построения углеродного скелета создаваемой молекулы. Между тем существует обширный класс синтетических задач, в которых необходимый скелетный каркас уже имеется в готовом виде, и для получения целевой структуры требуется лишь тем или иным путем изменить систему функциональных групп.
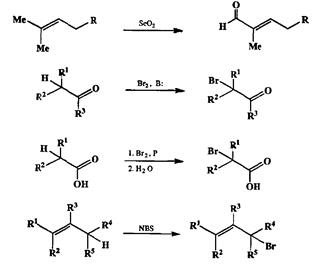 Схема 2.64
Схема 2.64
|
Дата добавления: 2015-04-05; просмотров: 1763;