Изогипсические трансформации. Синтетическая эквивалентность функциональных групп одного уровня окисления.
Как мы уже могли убедиться, функциями, наиболее часто возникающими при сборке связи С—С, являются спиртовая (реакции Гриньяра, альдольная конденсация) и олефиновая (реакция Виттига, кротоновая конденсация). К этому следует добавить, что именно к спиртам и алкенам приводят основные неизогипсические трансформации производных высших уровней окисления, такие, как восстановление карбонильных соединений и частичное гидрирование тройной связи. Поэтому неудивительно, что в ряду изогилсиче-ских превращений уровня окисления 1 ключевое место занимают реакции спиртов и алкенов.
С синтетической точки зрения важнейшими изогипсическими трансформациями спиртов являются образование алкилгалогенидов и сложных эфиров, включая эфиры сульфокислот. Это обусловлено тем, что производные этих типов широко используются как синтетические эквиваленты карбокатионов R+ для построения связи С-С (например, при алкилирова-нии енолятов). Реакции, приводящие к получению из спиртов простых или сложных эфиров или алкилгалогенидов, вообще говоря, относятся к числу тривиальнейших превращений. Поэтому может показаться довольно странным, что не прекращается поток публикаций, в которых описываются «новые и эффективные» методы осуществления этих превращений. Представление об интенсивности подобных поисков дает хотя бы тог факт, что в монографии Ларока [19с], целиком посвященной рассмотрению методов проведения разнообразных органических реакций, приводится более 40 методов для такого частного случая, как превращение спирта в соответствующий хлорид. Список используемых для этой цели реагентов включает как вполне тривиальные вещества, например НО или SOCt, так и более экзотические переносчики аниона С1~ типа Ph-sP/CCLi, Me3SiCl, (PhO)3P/PhCH2Cl и т.п. Столь же разнообразны и многочисленны процедуры, предлагаемые для других изогипсических превращений спиртов. Необходимость наличия столь обширного арсенала методов обусловлено прежде всего тем, что в ходе полного синтеза может понадобиться решение задачи проведения одного из этих простых превращений спиртовой функции в применении к субстрату, структура которого по каким-либо причинам не допускает использования обычных методов. Именно благодаря постоянно ведущейся интенсивной разработке как всевозможных вариантов старых методов, так и принципиально новых процедур, можно утверждать, что практически для любого случая удастся подобрать надежный способ осуществления превращений типа ROH-> ROAc (RHal, ROSO2R') независимо от особенностей общего структурного контекста.
Особенно важными для синтетической практики являются такие производные спиртов, как эфиры л-толуолсульфокислоты (тозилаты), а также эфиры метансульфо- и трифторметансульфокислот (мезилаты и трифлаты). Наличие в этих соединениях хорошей уходящей группы делает их практиче ски универсальными субстратами для осуществления широчайшего спектра изогипсических превращений на уровне окисления 1 по общей схеме нуклео-фильного замещения:
R-OSO2R1 + Nu- -> R-Nu + -OSO2R1
R« - л-МеС6Н4, Me, CF3; Nu - Hal, OR*, OCOR>, S.W, N(R*)2, N3> NO2 и т.д.
Алкилгалогениды, естественно, также могут служить субстратами в показанной выше реакции, причем в особенно важном ее варианте, а именно при получении фосфониевых солей R-P+Ph3 НаГ — предшественников для генерации соответствующих фосфоранов — реагентов Виттига. Отметим, что алкилгалогениды всевозможных структурных типов легко могут быть превращены в эквиваленты карбанионов — магний- или литийорганические производные, которые являются важнейшими синтетическими реагенты нулевого уровня окисления.
Изогипсические превращения, такие, как электрофильное присоединение воды, спиртов или галогеноводородов по двойной связи, часто используется для превращения алкенов в спирты, эфиры или алкилгалогениды соответственно. Первоначально область препаративной применимости этих превращений ограничивалась тем обстоятельством, что несимметричные ал-кены типа 125реагировали неселективно, образуя не только продукты присоединения по правилу Марковникова (М-аддукты), но и продукты присоединения против правила Марковникова (аМ-аддукты), а также продукты перегруппировок. Как уже отмечалось выше (схема 2.10), проблему селективного получения М-аддуктов, например 126(схема 2.47), удалось решить, разработав альтернативный протокол проведения реакции, включающий стадии сольвомеркурирования алкенов и восстановительного демеркурирования образующихся ртутьорганических аддуктов (схема 2.47).
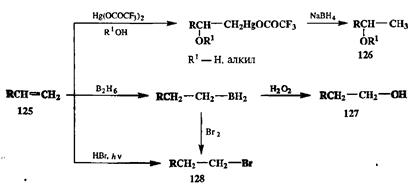 Схема 2.47
Схема 2.47
|
Столь же селективно может быть выполнено получение аМ-аддукта 127с помощью последовательности неизогипсических стадий — гидробори-рования (восстановления) и окисления. Отметим также, что, если на второй стадии использовать в качестве окислителя не пероксид водорода, а бром, то продуктом реакции будет аМ-бромид 128.Кстати, этот же продукт может быть получен непосредственно из алкена путем гомолитиче-ского присоединения бромоводорода.
Ранее мы уже приводили различные примеры реакций образования связей С-С, ведущие к получению алкенов с концевой двойной связью. Возможность селективного превращения таких алкенов в продукты, содержащие функциональные группы у тгрминального атома углерода (например, 127или 128),создает возможность реализации схемы последовательного удлинения углеродной цепи с использованием трех стандартных операций, а именно:
1) синтез алкена с терминальной связью;
2) превращение этого продукта в терминально функционализирован-ное производное типа 128;
3) использование последнего в одной из реакций, ведущих к образованию связи С—С.
Реакции обратного типа — образование двойной связи путем элиминирования HHal или HiO из соответствующих производных — широко употребляются в синтетической практике в самых различных ситуациях.
Очевидно, что синтетическая применимость подобного пути также более всего определяется возможностью обеспечения селективности элиминирования для случая несимметричных субстратов, что во многих случаях может быть достигнуто подбором условий проведения и варьированием природы реагентов (см. ссылки в соответствующих разделах монографии [19с]).
Учитывая все множество методов, разработанных для проведения разнообразнейших взаимопревращений между функциональными производными первого уровня окисления, можно считать эти функции синтетически эквивалентными. Это означает, что задача введения любой из них в данный фрагмент синтезируемой структуры может считаться успешно решенной, если в результате использования той или иной конструктивной реакции в этом фрагменте возникает, например, двойная связь или спиртовая функция.
На уровне окисления 2 рассмотрим прежде всего карбонильные соединения и ацетилены, легко получаемые как по многих реакциях образования связи С-С, так в результате неизогипсических трансформаций других функциональных групп.
Пожалуй, наиболее значимым типом изогипсических трансформаций этих соединений являются их превращения в синтетические эквиваленты карбанионов, как показано на схеме 2.48.
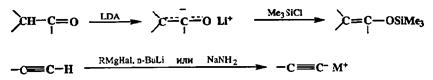 Схема 2.48
Схема 2.48
|
Роль образующихся при этом производных в построении связей С—С ранее уже обсуждалась достаточно подробно. Подчеркнем лишь, что, помимо указанных на схеме силиловых енолятов, в практике современного синтеза находят все большее применение ковалентные еноляты других элементов, например бора, титана, олова или циркония. Возможность использования подобных вариантов особенно важна для тех случаев, когда необходимо не только обеспечить высокую эффективность той или иной конденсации, но и надежно контролировать ее стерический ход.
Очень важным и в лабораторном, и в промышленном синтезе является присоединение к терминальным алкинам спиртов, карбоновых кислот и га-логеноводородов, приводящее к соответствующим винильным производным (схема 2.49).
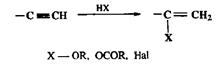 Схема 2.49
Схема 2.49
|
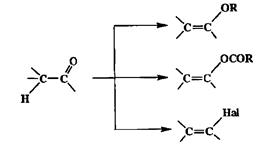
Эти соединения могут также рассматриваться как производные енолов, и во многих случаях в лаборатории оказьшается предпочтительным получать их из кетонов (схема 2.50).
|
| Схема 2.50 |
Первоначально интерес к соединениям этого типа, особенно винилгало-генидам, был в основном связан с их использованием в качестве мономеров в производстве различных пластических материалов. В настоящее время они также находят разнообразное применение в лабораторном синтезе и как синтетические эквиваленты винил-катионов (в реакциях с купратными производными в качестве нуклеофилов), и как предшественники для получения нуклеофильных реагентов типа виниллитиевых или винилмагниевьк производных, эквивалентов винил-анионов (схема 2.51).
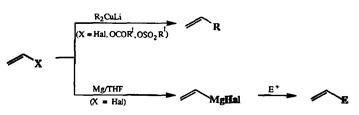 Схема 2.51
Схема 2.51
|
Ко второму уровню окисления, вообще говоря, относятся любые производные, содержащие две функциональные группы первого уровня окисления. Если эти функции достаточно удалены друг от друга, так, что они не могут оказывать заметного взаимного влияния, то тогда все, что говорилось о производных первого уровня окисления, применимо и к этим случаям. Иная ситуация возникает, когда такие функции находятся у соседних атомов углерода, так что по сути дела они образуют единую функциональную группу, как это имеет место в таких соединениях, как оксираны (эпоксиды), 1,2-дизаме-щенные (вицинальные) и аллильные производные. Эпоксиды и 1,2-бифунк-циональные производные алканов — это родственная группа соединений, внутри которой изогипсические трансформации легко осуществимы с помощью стандартного набора внутри- и межмолекулярного нуклеофильного замещения, как показано на схеме 2.52.
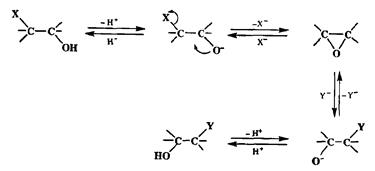 Схема 2.52
Схема 2.52
|
Направлением раскрытия оксиранового цикла можно управлять за счет юриаций условий реакции и природы нуклеофила, что позволяет в необхо-шмых случаях осуществлять обмен местами соседних функциональных групп (через стадии замыкания и раскрытия эпоксидного кольца), а также обеспечивать нужную конфигурацию включенных в такие системы асимметрических центров.
Основной путь синтеза эпоксидов и вицинальных бифункциональных производных — неизогипсические трансформации олефинов, о которых мы скажем несколько ниже. Эпоксиды могут быть получены также как непосредственные продукты реакций при использовании таких методов создания связи С-С, как реакция Дарзана (модификация классической конденсции альдольного типа, в которой в качестве метиленовой компоненты используются а-хлорэфиры 130),или присоединение илидов серы, например диме-тилсульфонийметилида (131),по карбонильной группе (схема 2.53). Оба эти метода основаны на легкости внутримолекулярного нуклеофильного замещения в первоначально образующихся вицинально замещенных интермедиатах (130аи 131асоответственно).
Среди изогипсических превращений производных второго уровня окисления особое место занимают превращения аллильных производных 132а и 132Ь(схема 2.54). Нуклсофильное замещение группы X в этих соединениях протекает с особой легкостью, и оно может идти как по месту нахождения этой группы, так и с аллильной перегруппировкой. Во многих случаях удается обеспечить надежный контроль за направленностью реакции и тем самым высокую селективность ее протекания по тому или иному маршруту. Поэтому изомерные производные 132аи 132Ьна самом деле справедливо рассматривать как синтетически эквивалентные.
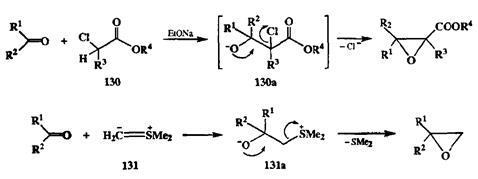 Схема 2.53
Схема 2.53
|
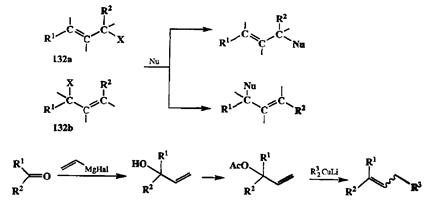 Схема 2.54
Схема 2.54
|
Повышенная склонность третичных аллильных производных претерпевать аллильную перегруппировку в ходе нуклеофильного замещения позволила разработать один из самых надежных способов удлинения углеродной цепи, ключевыми стадиями которого являются: а) синтез третичного аллиль-ного карбинола из кетона по обычной схеме реакции Гриньяра, б) превращение гидроксильной функции в более легко уходящую группу (например, ацилокси) и в) сочетание полученного производного с алкиллитийкупрат-ным реагентом (см. схему 2.54).
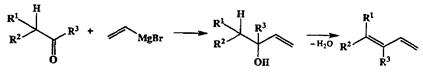 Схема 2.55
Схема 2.55
|
Другое важное изогипсическое превращение аллильных производных — элиминирование группы Н-Х, приводящее к 1,3-диенам. Помимо того, что многие представители этой группы соединений практически важны как мономеры, 1,3-диены занимают особое место в органическом синтезе как субстраты для реакции Дильса—Альдера. Один из самых обычных путей синтеза этих производных основан на последовательности превращений, влю-чающих стадию реакции Гриньяра карбонильных соединений с винильными производными с последующим элиминированием воды (схема 2.55) (иногда предпочтительнее сначала превратить аллильный спирт в соответствующее ацетоксипроизводное).
Помимо методов, включающих образование новых связей С—С (реакций Гриньяра и некоторых других), для получения аллильных производных можно использовать еще ряд чисто трансформационных превращений. Некоторые из них, например, восстановление а, р-непредельных карбонильных соединений (обычных продуктов конденсации кротонового типа), аллильное галогенирование алкенов с помощью N-бромсукцинимида (NBS) и изомеризация эпоксидов в аллильные спирты под действием триметилсилилтриф-лата {19d], показаны на схеме 2.56.
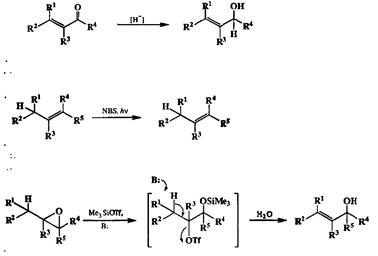 Схема 2.56
Схема 2.56
|
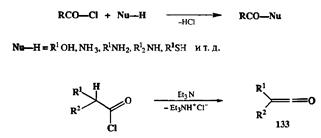 Схема 2.57
Схема 2.57
|
На третьем уровне окисления важнейшие производные, от которых с помощью изогипсических трансформаций возможен переход «к чему угодно», — это галогенангидриды, легко получаемые из карбоновых кислот по хорошо отработанным методикам. Галогенангидриды, как мы уже отмечали ранее, являются электрофилами, синтетическими эквивалентами ацил-катионов RCO+. В этой роли они используются как для ацилирования по гегероатому таких соединений, как спирты, амины или тиолы (схема 2.57), так и для С-ацилирования по реакции Фриделя—Крафтса. Специфической особенностью хлорангидридов является их способность под действи ем третичных аминов претерпевать изогипсическое превращение в кетены 133— реагенты, широко применимые для [2 + 2]-циклолрисоединения (см. разд. 2.6.3.2).
Среди бифункциональных производных третьего уровня окисления следует прежде всего упомянуть пропаргиловые спирты (этинилкарбинолы). Их получение по реакции нуклеофильного присоединения ацстиленидов по карбонильной группе альдегидов и кетонов — это настолько хорошо отработанный метод, что можно уверенно относить пропаргиловные спирты 134любых структурных типов к разряду легко доступных соединений. Один из важнейших путей их препаративного использования основан на реакции соответствующих ацетатов с купратными реагентами, которая протекает с про-паргил-алленильной перегруппровкой и служит надежным методов получения алленов 135.Среди множества других превращений следует выделить прежде всего показанную на схеме 2.58 последовательность двух изогипсиче-ских превращений 134,а именно: элиминирования воды, приводящего к 1,3-енинам, и гидратации последних, в результате чего образуются а, р-непре-дельные кетоны 136.
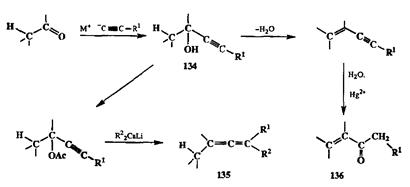 Схема 2.58
Схема 2.58
|
К тому же типу бифункциональных производных третьего уровня окисления относятся а, р-непредельные альдегиды и кетоны. Ранее мы уже говорили об их роли в таких синтетически важных реакциях, как реакции Дильса—Альдера или Михаэля (см. разд. 2.3). Важнейшие изогипсические трансформации этих производных основаны на их способности легко присоединять самые различные ггтероатомные нуклеофилы по кратной утлерод-утле-родной связи, что делает доступным широкий набор р-замещенных функциональных производных карбонильных соединений (а из них и соответствующих спиртов). Отметим, что в большинстве случаев получаемые аддукты способны претерпевать также обратное превращение — элиминирование фрагмента H-Nu (схема 2.59).
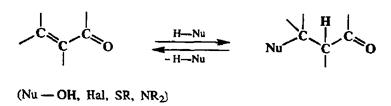 Схема 2.59
Схема 2.59
|
Дата добавления: 2015-04-05; просмотров: 2088;