Карбокатионные или карбанионные реагенты. О некоторых дополнительных возможностях проведения реакций образования связи С-С
Вначале разд. 2.2.3 мы не делали никаких принципиальных различий между карбокатионами и карбанионами, рассматривая и те, и другие в качестве равноправных партнеров в гетеролитических реакциях образования связей С—С. Однако читатель мог заметить, что на самом деле в реальных синтетических методах, о которых до сих пор шла речь, такого равноправия нет. Действительно, мы видим, что в этих реакциях в роли нуклеофилов могли использоваться либо карбанионы как таковые (ионные еноляты, ацетиле-ниды, илиды), либо приближающиеся к ним по свойствам высокополяризо-ванные реагенты типа литий- или магнийорганических производных. Напротив, синтетическими эквивалентами карбокатионов, как правило, служили чисто ковалентные электрофилы. Мы почти не рассматривали обратную ситуацию, в которой электрофилом был бы «живой» карбокатион как активный реагент, а нуклеофилом — некий ковалентный эквивалент карбаниона. Подобная «асимметрия» подхода вовсе не случайна. Она является отражением того, что карбанионы — более стабильные частицы и их легче генерировать и использовать [4, 8], чем карбкатионы. В карбанионах углеродный атом, несущий заряд, имеет заполненный октет электронов, ему, так сказать, «ничего больше не нужно», в силу чего в целом для этих ионов гораздо менее характерно протекание побочных реакций типа скелетных перегруппировок, что, напротив, свойственно карбокатионным частицам [3]. Поэтому эффективная стабилизация карбанионов как реагентов и интермедиатов относительно легко может быть обеспечена такими способами, как использование соответствующих растворителей, противоионов или лигандов или, наконец, путем введения дополнительных электроноак-цепторных групп в молекулу предшественника. В то же время достичь такой цели в применении к электрофильным реагентам гораздо труднее (хотя известно немало способов повышения стабильности карбокатионов, но они, как правило, менее удобны и имеют достаточно ограниченную область применения) [3, 17а].
В силу сказанного становится понятным, почему большинство классических синтетических методов, описываемых в терминах ионных реакций, основаны по сути дела на одной и той же общей схеме сочетания: ионный нуклеофил + ковалентный электрофил, а не на альтернативном варианте: ионный электрофил + ковалентный нуклеофил. Очевидным исключением в этом отношении является электрофильное замещение в ароматическом ряду (реакция Фриделя-Крафтса), в которой именно карбокатионные реагенты выступают в роли электрофилов, а нуклеофилами служат ковалентные ароматические субстраты. При этом следует отметить, что жесткость классических условий проведения алкилирования или ацилирования по Фриделю— Крафтсу делают этот метод малоприменимым по отношению к кислотола-бильным субстратам, и поэтому использование этой реакции в полном синтезе ограничено. Между тем за последние 10—15 лет все большее внимание уделяется развитию новых эффективных и общих методов стабилизации карбокатионов как реагентов и интермедиатов, и к настоящему времени уже накоплено достаточно данных, позволяющих утверждать, что синтетические методы, основанные на реакциях ионных электрофилов с ковалентньши нуклеофилами, могут явиться существенным дополнением к уже существующим традиционным методам образования связи С-С с помощью карбани-онньгх реагентов. Рассмотрим некоторые примеры, иллюстрирующие это утверждение.
Ранее мы уже отмечати, что классическая реакция Гриньяра может быть описана формальной схемой сочетания анионоподобного нуклеофила с ко-валентным (хотя и поляризованным) электрофилом. На схеме 2.41 приведены примеры успешного использования для тех же целей альтернативного подхода, основанного на взаимодействии ковалентного нуклеофила, аллил-силана 109а или 109Ь,с катионоподобным электрофилом, образуемым из соответсвующих карбонильных соединений (или их производных) под действием кислот Льюиса (17Ь,с]. Если же в качестве нуклеофилов в такой реакции использовать силиловые эфиры енолов, например 110,то результатом сочетания в этом случае будет образование аддуктов альдольного типа (реакция Мукаямы [17d]).
Как аллилсиланы, так и силиловые эфиры енолов являются отличными нуклеофилами для реакций со стабилизированными карбокатионами и иных типов, помимо тех.что показаны на схеме 2.41. На схеме 2.42 приведены примеры алкилирования этих субстратов третичными карбокатионами, такими, как 111или 112,генерируемыми соответственно из 1-хлорадамантана или 1-метил-1-хлорциклогексана [17с,е1. Кстати можно сказать, отвлекшись на минуту от карбокатионной темы, что некоторые силильные производные, например, силилкетекацетали, способны реагировать в высокополярных растворителях со столь сильными электрофилами, как некоторые акцепторы Михаэля, даже в отсутствие кислот Льюиса [17b,f,g|. Примером этою может служить реакция циклопентенона (113)с силилкетенацеталем (114)[17f].
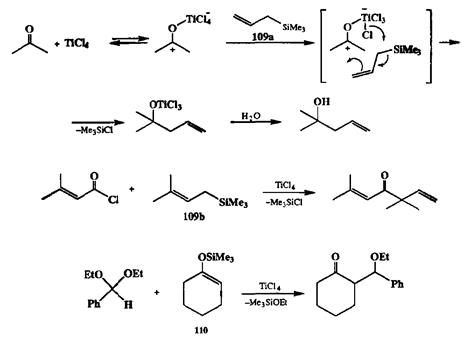 Схема 2.41
Схема 2.41
|
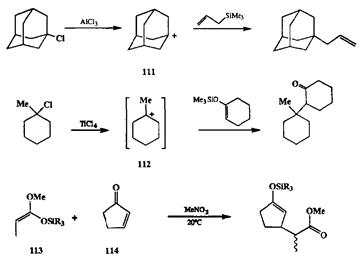 Схема 2.42
Схема 2.42
|
Показанная на схемах 2.41 и 2.42 возможность реализации превращений того же типа, что был описан ранее, но с помощью реагентов иных классов и в совершенно других условиях, позволяет резко расширить границы применения многих синтетически важных реакций. Так, например, хорошо известное алкилирование ионных енолятов великолепно работает, если в качестве алкилирующих реагентов используются первичные алкилгалогенвды (или производные первичных спиртов). Однако эта реакция абсолютно неприменима для тех случаев, когда в качестве электрофилов берутся третичные гало-гениды, поскольку последние немедленно подвергаются дегидрогалогениро ванию под действием ионных енолятов как оснований. Эти осложнения полностью устраняются, если использовать показанный на схеме 2.42 вариант алкилирования ковалентных енолятов ионными электрофилами. В реакциях этого типа получение карбонильных производных, содержащих трет-ал-кильный заместитель у а-углеродного атома, не составляет какой-либо проблемы [17е]. Совершенно новые синтетические возможности открылись при проведении конденсаций альдольного типа по схеме взаимодействия ковалентных енолятов с карбонильными производными, активированными кислотами Льюиса (см. пример на схеме 2.41). Основное преимущество этого варианта связано с тем, что он дает возможность в широких пределах контролировать стереохимию сочетания путем варьирования природы ковалент-ного енолята и/или кислоты Льюиса [17g] — возможность, практически недостижимую для классических условий проведения альдольной конденсации. Собственно, именно на основе этого подхода удалось решить неимоверно сложную задачу контроля стерическогохода реакций альдольной конденсации на всех стадиях синтеза аддукта 74 (см. схему 2.27).
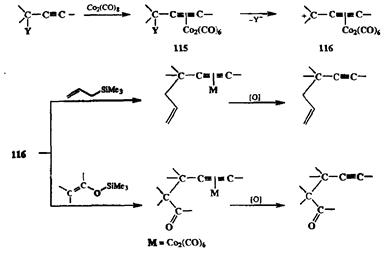 Схема 2.43
Схема 2.43
|
В последние десятилетия сложилось новое направление в химии карбока-тионов, основанное на способности комплексов переходных металлов стабилизировать положительный заряд на соседнем атоме углерода. Пожалуй, наиболее яркой иллюстрацией эффективности такой стабилизации могут служить результаты исследований, посвященных химии дикобалътгексакарбонильных (ДКГК) комплексов ацетиленовых производных. Начало этим исследованиям было положено в работах Николаса, который обнаружил, что ДКГК-комплексы пропаргиловых спиртов общей формулы 115 (Y=OH, схема 2.43) могут быть легко превращены при действии сильных кислот в ДКГК-комплексы соответствующих пропаргильных катионов типа 116, некоторые из которых стабильны не только в растворах, но и могут быть выделены в свободном состоянии.
Этот факт был интересен сам по себе, поскольку до этого образования пропаргильных катионов как стабильных частиц не удавалось наблюдать даже при попытках их генерации при низкой температуре в растворах суперсильных кислот (условия «долгой жизни» для карбокатионов самого различного строения). С точки зрения синтеза особенно важным явилось то, что карбокатионы 116 оказались весьма активными электрофилами, легко вступающими в реакции с различного рода С-нуклеофилами, такими, как аллилсиланы или силиловые эфиры енолов [18а}. Окислительная декомплексация получаемых при этом аддуктов (см. реакции на схеме 2.43) приводила к конечным продуктам превращения, образование которых в точности соответствовало формальной схеме сочетания пропаргильного катиона с ковалентным нуклеофилом. Поскольку пропаргиловые спирты, первичные, вторичные или третичные, почти любого строения относятся к разряду легко получаемых производных, а их превращения типа показанных на схеме 2.43 протекают, как правило, гладко и с хорошими выходами, неудивительно, что использование ДКГК.-комплексов алкинов в реакциях электрофильного пропаргилирования различных субстратов (реакция Николаса) является сейчас одним из стандартных методов в синтетической практике. В связи с этим уместно подчеркнуть, что прежние методы, основанные на использовании пропаргилгалогенидов в качестве электрофилов, имели очень ограниченную область применения, поскольку в таких реакциях могли использоваться лишь первичные пропаргилгалогениды, и к тому же в ряде случаев наблюдалось образование побочных продуктов из-за , возможности ацетилен–алленовой перегруппировки. При проведении пропаргилирования по схеме реакции Николаса этих ограничений просто не существует.
Возможность обеспечить эффективную стабилизацию карбокатионных интермедиатов в комплексах типа 116 позволила реализовать новый вариант проведения электрофильного присоединения по двойной связи сопряженных енинов 117 в виде последовательности кинетически независимых стадий присоединения электрофила и нуклеофила, как показано на схеме 2.44. Исходным субстратом в этой реакции является ДКГК-комплекс 118, а ключевым звеном — образование стабильного катионоидного ин-термедиата 119 в результате присоединения катионоидного электрофила по двойной связи. Этот интермедиат также оказался способным вступать в реакции с рядом типичных ковалентных нуклеофилов, и таким образом в результате последовательности операций, проводимых в одном реакционном сосуде, удалось получить (после окислительной декомплексации аддуктов 120) набор структурно различных продуктов типа 121. Примечательно, что как природу исходного сопряженного енина, так и природу электрофильного и нуклеофильного компонентов показанного сочетания можно варьировать совершенно независимо и в довольно широких пределах.
Показанная здесь последовательность реакций отражает сборку конечной структуры из трех сравнительно некрупных фрагментов. Ранее мы уже рассматривали подобного рода сборку, основанную на реакции нуклеофиль-ного присоединения (см. схему 2.31), и нетрудно заметить «зеркальную» аналогию в этих двух подходах. В самом деле, в обоих случаях речь идет о гете-ролитическом присоединении по кратной связи. Эти реакции, будь то элект-рофильные или нуклеофильные, состоят в присоединении к субстрату двух аддендов противоположного знака, что можно описать следующей общей схемой:
субстрат + Е+ + Nu" → Е-субстрат—Nu
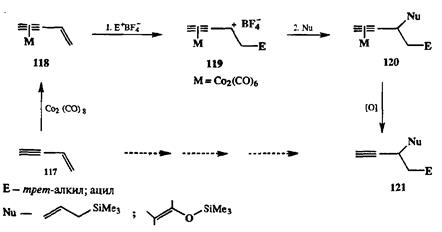 Схема 2.44
Схема 2.44
|
Последовательность присоединения (сначала электрофил, затем нук-леофил или наоборот) определяет принадлежность реакции к классу электрофильного или нуклеофильного присоединения и, соответственно, протекание реакции через образование катионоидного или анионоидного интермедиатов. В традиционных методах проведения подобных реакций природа второго (по очередности присоединения) адденда изначально жестко задана структурой реагента и/или составом реакционной среды. Так, скажем, нуклеофильное присоединение (классическая реакция Михаэля) предполагает нейтрализацию карбанионного интемедиата протоном из среды, а электрофильное присоединение завершается взаимодействием катионоидного интермедиата либо с противоионом, имеющимся в составе исходного электрофила, либо с каким-либо нуклеофилом, присутствующим в среде. В современных вариантах проведения этих реакций по схеме последовательного присоединения обеспечивается возможность образования карбанионных или карбокатионных интермедиатов как кинетически независимых частиц, что и позволяет независимо варьировать природу обоих аддендов, и таким образом производить сборку целевой молекулы из трех «кубиков», выбираемых по желанию синтетика из обширного набора возможностей.
Очевидно, что наличие двух «зеркальных» вариантов подобного последовательного присоединения (ионный нуклеофил + субстрат, затем электрофил или ионный электрофил + субстрат, затем нуклеофил) резко расширяет традиционные рамки синтетического применения гетеролитиче-ского присоединения и поднимает эти реакции до уровня стратегически значимых.
Дата добавления: 2015-04-05; просмотров: 1703;