Классификация проблем селективности
Мы уже неоднократно говорили о селективности тех или иных органических реакций. Тем не менее, этот вопрос настолько важен для органического синтеза в целом, что он заслуживает более легального обсуждения как самостоятельная проблема.
Надежность синтетического метода не только предполагает, что данный метод может использоваться для эффективного осуществления требуемого превращения, но и подразумевает, что в избранных условиях между данной функциональной группой и реагентом протекает одна и только одна реакция. Тем не менее, этим проблема селективности далеко не исчерпывается. Дело в тем, что реальный субстрат может содержать несколько одинаковых или близких по свойствам функциональных групп, способных реагировать с одним и тем же реагентом, а по условиям задачи требуется провести превращение с одной из них, Кроме того, даже при наличии всего лишь одной функциональной группы, се превращение с использованием «чистой» (т.е. надежной) реакцииможет приводить к образованию нескольких изомерных продуктов. Характер проблем, связанных с селективностью, может быть весьма различен. Ниже мы рассмотрим некоторые типичные случаи, с которыми чаще всего приходится иметь дело в рамках решения задач обеспечения селективности тех или иных превращений. Если взглянуть на проблему селективности с точки зрения кинетики, то можно выделить три общих типа случаев, в каждом из которых возможно образование более, чем одного продукта в условиях данной реакции.
Тип 1. Последовательные реакции. На схеме 2.68 приведен ряд примеров таких последовательностей, в которых продукт, образующийся в результате первой реакции, способен подвергаться дальнейшему превращению в той же реакционной системе. Следовательно, в этом случае для достижения селективности требуемого превращения необходимо иметь возможность остановить процесс на какой-либо из стадий.
 Схема 2.68
Схема 2.68
|
Этого можно добиться разными способами. Например, обе реакции в превращении (1) принадлежат к одному и тому же типу гидрирования в присутствии гетерогенного катализатора. Поэтому для обеспечения селективного гидрирования ацетиленов в олефины необходимо модифицировать катализатор так, чтобы восстановление двойной связи на этом катализаторе проходило существенно медленнее, чем восстановление тройной. Этому требованию отвечает, например, катализатор Линдлара — палладий, осажденный на карбонате кальция и дезактивированный добавками оксида свинца (Pd-СаСОз—РЬО).
Напротив, стадии окисления первичных спиртов в альдегиды, а последних в кислоты [последовательноть (2)| резко различаются по своему механизму, что позволяет осуществить первую из этих реакций селективно за счет использования специфических реакций и реагентов. Для этой цели, например, очень эффективна система ДМСО — кислота Льюиса (см. схему 2.60), не способная окислять альдегиды.
Обеспечение селективности алкилирования енолятов [превращение (3)] — это одна из центральных проблем в синтетической химии карбонильных производных, для решения которой разработан комплекс разнообразных приемов, которые рассмотрены в разд 2.4.4.
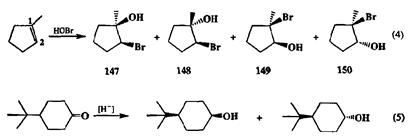 Схема 2.69
Схема 2.69
|
Тип 2. Параллельные реакции. В примерах, показанных на схеме 2.69, смеси однотипных продуктов образуются благодаря наличию нескольких конкурирующих каналов для данной реакции, и в этих случаях для достижения селективности превращения требуется обеспечить условия для преимущественного (а лучше исключительного) протекания реакции по требуемому направлению. В случае (4) конечный результат превращения зависит, во-первых, от направления атаки Вг4" на тот или иной атом углерода двойной связи, что определяет соотношение позиционных изомеров в образующейся смеси (147+ 148): (149+ 150),а во-вторых, от ориентации подхода нук-леофила НО", определяющей образование продуктов цис- или транс-прн-соединения [соотношение (147+ 149): (148+ 150)).Если селективность образования продуктов присоединения по правилу Марковникова, 147и 148,— это легко достижимая цель, то обеспечение эффективного управления стереохимией присоединения уже относится к категории довольно непростых задач. В сходном случае [(реакция (5)] стереохимия продукта восстановления определяется направлением атаки гидридного реагента по карбонильной группе с одного из двух альтернативных направлений — «снизу» или «сверху» плоскости цикла. Принципы решения этой проблемы обсуждены в разд. 2.4.3.
Тип 3. Последовательно-параллельные реакции. Для примеров, приведенных на схеме 2.70, характерны трудности и первого, и второго типов превращений. Поскольку показанные субстраты полифункциональны, то уже пер-вая из стадий может приводить к образованию смесей продуктов реакций. Доплнительное осложнение обусловлено тем, что каждый из образующихся при этом продуктов, в свою очередь, способен реагировать с тем же реагентом.
Очевидно, что задача обеспечения селективности реакции в таких превращениях существенно сложнее, чем в рассмотренных выше случаях. Действительно, для избирательного получения одного из продуктов первой стадии необходимо не только обеспечить селективное протекание этой реакции по одной из имеющихся групп, но и заблокировать следующие стадии, г. е. обеспечить инертность первичного продукта в условиях реакции. Здесь могут использоваться самые различные приемы. Так, например, при ацети-лировании глицерина [превращение (6)] задача селективного получения мо-но- или бисацетилированных производных по первичной гидроксильной группе (группам) сравнительно легко решается при использовании такого мягкого реагента, как уксусный ангидрид, в требуемых стехиометрических количествах. В то же время моноацетилирование вторичного гидроксила достижимо лишь при условии, что оба первичных гидроксила защищены какой-либо легко удаляемой впоследствии группой (подробнее о принципах использования защитных групп см. разд. 2.4.5.). Дополнительные трудности возникают в тех нередких случаях, когда продукт, получаемый на первой стадии, оказывается более реакционноспособным, чем исходный субстрат. Так, например, при алкилировании толуола по Фриделю—Крафтсу [реакция (7)) присоединение первой алкильной группы резко повышает нуклеофильность ароматического ядра, так что повторное алкилирование протекает быстрее, чем первая стадия. Взаимное влияние функциональных групп является отнюдь не исключением, а правилом, особенно в тех случаях, когда функциональные группы сближены или разделены системой кратных связей. Однако подобное влияние может иметь результатом не только ускорение, но и замедление реакций. Именно этим можно воспользоваться для того, чтобы добиться моноалкилирования толоула. С этой целью вместо аликлирования используют ацилирование, при котором входящая ацильная группа пассивирует ароматическое ядро по отношению к электрофильной атаке. Благодаря этому реакция протекает почти исключительно как монозамещение. Последующее восстановление
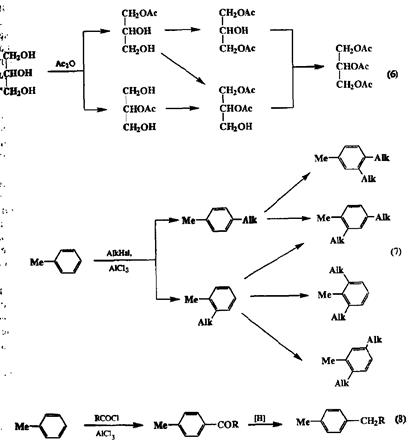 Схема 2.70
Схема 2.70
|
кетогруппы в полученном продукте и даст требуемое моноалкилпроиззодное толуола [реакция (8), схема 2.70].
Рассмотренные типы ситуаций ясно показывают, сколь многогранна и сложна проблема селективности в целом. Вообще говоря, любое органическое соединение полифункционально (даже простейшее из них — метан — образует при хлорировании набор продуктов от СН3Сl до ССl4)- Поэтому неудивительно, что проблема селективности реакций является в действительности ключевой при планировании синтеза.
Из нашего схематического рассмотрения видно, сколь различным может быть характер препятствий на пути к достижению желаемого результата. Соответственно, различны и принципы решения синтетических задач на селективность. Возможность решения задач, связанных с возможностью протекания параллельных реакций (тип 1), в значительной степени обусловлена такими необходимым характеристиками синтетического метода, как его чистота и избирательность. В самом общем виде эти вопросы мы уже обсуждали в предыдущих разделах. Поэтому ниже мы сосредоточим внимание главным образом на задачах, относящихся к типу 2 и — в меньшей степени — к типу 3 по нашей классификации. Речь пойдет главным образом о некоторых принципах решения задач, основанных на вариациях в природе реагента, структуры субстрата и химизма основной реакции. Стоит отметить, что эти пути решения, хотя они и относятся к категории основных, отнюдь не являются единственными. Определенную пользу может принести также и чисто физический приемы, такие, как удаление целевого продукта из равновесной смеси или управление ходом реакции, основанное на понимании кинетических закономерностей конкурирующих процессов [22а].
Теперь несколько терминологических замечаний. Предпочтительное протекание реакций по одной из нескольких родственных, но химически различных функциональных групп сусбетрата, обычно называют хемоселективностью. Если речь идет об избирательности по отношению к определенному положению в молекуле, принято говорить о региоселективности. Если же имеется в виду предпочтительное образование одного из пространствен-(ных изомеров, то пользуются термином стереоселективность. Наконец, гели удается добиться полной селективности, то такой результат характеризуют Термином специфичность (соответственно хемо-, регио- или стерео-). Наконец, существует еше один аспект селективности, связанный с возможностью образования двух оптических антиподов — энантиомеров. Обсуждению общих подходов к решению этой чрезвычайно важной проблемы, а также методологии разработки частных ее решений посвящено несколько десятков монографий и сотни обзоров (см., например, [22Ь]). Как нам представляется, вряд ли было целесообразно пытаться дать содержательное изложение результатов поисков в этом направлении в контексте обсуждения данной главы. С некоторыми общими принципами, применяемыми для решения проблем энантиоселективности, читатель сможет ознакомиться в гл. 4 (см. разд. 4.2.3.3).
Дата добавления: 2015-04-05; просмотров: 2387;