Бадмаев Б.Ц. 7 страница
• типичный, положительно реагирующий на специфическую антисыворотку к острому лимфобластному лейкозу (ОЛЛ). Составляет 75 % ОЛЛ;
• Т-клеточный. Даетрозеткообразование. Встречается в 10 % случаев;
• В-клеточный. Имеет иммуноглобулины G на поверхности. Встречается менее чем в 5 % случаев;
• нуль-клеточный. Содержит терминальную нуклеотид-трансферазу. Встречается в 10 % случаев.
Некоторые из лимфобластных лейкозов имеют определенные хромосомные маркеры:
• ОЛЛ-Ь2 имеет уникальный хромосомный дефект t(4; 11) (q21 ;q23);
• ОЛЛ-ЬЗ имеет дефект хромосом — t(8,14)(q24.1 ;q32.3);
• ОЛЛ-U — имеет дефект хромосом — t(9,22)(q34.1 ;q11.2). Нелимфобластные (миелогенные) лейкозы подразделяются
морфологически на 6 подгрупп:
• М1 — незрелые миелобласты; до 3 % пероксидазо-положительные, могут иметь азурофильные гранулы и тельца Ауэра;
• М2 — зрелые миелобласты, более 50 % составляют промиелоциты. Встречаются тела Ауэра. Первые два типа составляют более 60 % этого вида лейкозов;
• МЗ — промиелоцитарный лейкоз. Характеризуется гипергрануляцией цитоплазмы с большими гранулами и множеством телец Ауэра. Ядро может быть многодольчатым. Составляет около 5 % данного типа лейкемий;
• М4 — миеломонобластный. Моноциты и промоноциты составляют до 20 %. Частота данного типа также в пределах 20 %;
• М5 — монобластный. Составляет менее 5 % случаев. Эти два типа (М4, М5) характеризуются повышенным содержанием лизоцима в сыворотке;
• Мб — эритролейкоз. Более 50 % клеток состоит из причудливых по форме предшественников эритроидного ряда. Составляет менее 5 % случаев.
Среди нелимфобластных миелогенных лейкозов также отмечаются хромосомные нарушения:
• ОМЛ-М2 имеет уникальные хромосомные отклонения — t(8;21) (q22.1;q22.3);
• ОМЛ-МЗ имеет уникальные нарушения хромосомных структур t(15;17)(q22;q11.2);
• ОМЛ-М4 имеет уникальное нарушение хромосом inv(16)(p13.2;q22). Кроме этого, описаны иные хромосомные нарушения, типичные для
нескольких типов ОМЛ одновременно. Так, отклонение типа +8 отмечается при ОМЛ М1, М2, М4, М5, Мб.
По качеству клеток в периферической крови в момент диагностики выделяются следующие типы:
• лейкемический,
• сублейкемический,
• алейкемический.
По характеру клинических проявлений лейкозы можно подразделять в зависимости от преобладающего клинического синдрома:
— интоксикационного,
— геморрагического,
— гиперпластического (лимфаденопатия, гепато- и спленомега- лия, экстранодулярный опухолевый рост),
— анемического,
— синдрома некротических изменений слизистых оболочек,
— синдрома нейролейкемии,
— желтушного синдрома,
— синдрома мочекислого диатеза.
Хронические миелопролиферативные лейкозы. Они характеризуются пролиферацией зрелых или способных к дозреванию клеток крови.
Пролиферация миелоидного ростка подразделяется следующим образом:
• Bcr-abi-позитивные лейкозы (имеющие филадельфийскую хромосому), — истинный хронический миелолейкоз;
• Bcr-аЫ-негативные лейкозы (не имеющие филадельфийской хромосомы).
Хронические лейкозы подразделяются на типичные и нетипичные. К типичным относятся:
• истинная полицитемия;
• эссенциальная тромбоцитопения;
• агиогенная миелоидная дисплазия — остеомиелофиброз.
К нетипичным:
• атипичный хронический миелолейкоз;
• хроническая нейтрофильный лейкоз;
• хронический базс?фильный — тучноклеточный лейкоз;
• хронический эозинофильный лейкоз.
По количеству клеток в периферической крови в момент диагностики лейкозы можно подразделять на следующие типичные варианты: лей- кемический, сублейкемический, вариант бластного криза с указанием цитоморфологической особенности бластов.
По стадиям развития лейкоза выделяются следующие:
• начальная, стадия развернутых клинико-гематологических проявлений,
• стадия клинико-гематологической компенсации,
• стадия трансформации в острый лейкоз.
По гематологической формуле различают типичный вариант (в лей- кограмме преобладают гранулоциты различной степени зрелости), ба- зофильный, эозинофильный.
Эритремия (полицитемия, болезнь Вакеза) имеет различные особенности. По стадиям различают:
• развернутая (эритремическая) без миелоидной метаплазии селезенки;
• развернутая (эритремическая) с миелоиднрй метаплазией селезенки,
• стадия исходов в острый лейкоз, хронический миелолейкоз, остео- миелофиброз, гипопластическое состояние кроветворения.
По клиническим проявлениям:
• плеторический,
• гепатоспленомегалический,
• гипертонический,
• тромботический,
• геморрагический,
• тромбогеморрагические варианты.
Для всех лейкозов характерны осложнения: мочекислый диатез (подагра, развивается вследствие разрушения клеток и выхода их содержимого в кровь), а также поражения внутренних органов.
Идиопатический остеомиелофиброз (остеомиелосклероз) с миелоидной метаплазией.
Различают следующие морфологические стадии:
• неравномерная пролиферация клеток трех ростков, особенно ме- гакариоцитов (I стадия);
• постепенное присоединение миелофиброза (II стадия);
• развитие остеомиелосклероэа (III и IV стадии).
Хронический моноцитарный лейкоз характеризуется разрастанием пула моноцитарных клеток в крови и тканях.
По клиническим проявлениям различают следующие варианты: анемический, гиперпластический (с увеличением селезенки и/или печени).
По количеству клеток в периферической крови определяют: сублейкемический и лейкемический варианты.
Хронические лимфопролиферативные заболевания характеризуются разрастанием клеток, внешне неотличимых от лимфоцитов здорового организма. Их подразделяют на лейкозы и лимфомы в зависимости от их распространенности. В тех случаях, когда они находятся в крови и костном мозге, ставится диагноз лейкоза. В тех случаях, когда в крови этих клеток немного и их образования представлены в виде узлов, ставится диагноз лимфомы. Эти подразделения довольно условны, т.к. обнаружить увеличение лимфоидной массы в лимфатических узлах или тканях можно при любом лимфолейкозе. Точно так же и при опухолевых лимфомах поражение костного мозга, а вместе с ним и крови встречается очень часто. Все различия определяются лишь выраженностью этого феномена. Предполагается наличие бесчисленного множества лимфом, определяемого тем лимфоцитарным клоном, который пролиферирует. Тем не менее, традиционный консерватизм в подразделении опухолей лимфоидной ткани до сих пор присутствует.
Хронические лимфолейкозы характеризуются пролиферацией неопластических лимфоидных клеток, которые чаще всего представлены В-клетками, неспособными к дальнейшей дифференциации в продуцирующие иммуноглобулины плазматические клетки, а также опухолевым разрастанием иных клеток лимфоидного ряда (Т-лимфоциты различных классов — Т4+, Т8+, лимфоциты — естественные киллеры и пр.) клетки распространяются по костному мозгу, лимфатическим узлам, селезенке, печени, выходят в кровь, могут образовывать скопления в других органах. Количество лейкоцитов в периферической крови может достигать очень больших цифр —более 500—600x109/л. Пролиферирующие лимфоциты внешне неотличимы от нормальных, но обладают повышенной способностью к разрушениям от механического воздействия. В связи с этим в мазках крови при микроскопии часто обнаруживают специфические пятна — тени Боткина—Гумпрехта.
Пролиферация лимфоидных клеток, способных продуцировать М-иммуноглобулины, определяет развитие заболевания, получившего название по имени описавшего ее автора, макроглобулинемии Вальденшт- рема. Этот вариант лимфолейкоза, при котором имеет место инфильтрация пролиферирующими лимфоцитами костного мозга, лимфоузлов и селезенки, так же как и крови имеет свои особенности в виде гипервискозного синдрома и повышенной кровоточивости. Последнее определяется дефектом тромбоцитов, ингибируемых вырабатываемым лимфоцитами белком — макроглобулином. Описаны заболевания, когда пролиферирующие неопластические лимфоидные клетки способны вырабатывать отдельные части молекулы иммуноглобулинов — различные
виды легких (каппа или лямбда) и тяжелых цепей. Эти заболевания получили названия болезни легких цепей, болезни тяжелых цепей.
Лимфопролиферативные заболевания, протекающие без выхода лимфоцитов в периферическую кровь, получили название неходжкинс- кихлимфом в отличие от болезни Ходжкина — лимфогранулематоза. Для заболеваний характерны распространенность патологического процесса, а также существование различий в лимфоидных клетках, составляющих опухолевую массу. Приводим наиболее распространенную классификацию неходжкинских лимфом.
Низкодифференцированные лимфомы:
• мелколимфоцитарная лимфома,
• мелколимфоцитарная плазмоцитоидная лимфома,
• фолликулярная мелкоклеточная лимфома с расщепленным ядром. Лимфомы промежуточной дифференциации: диффузная крупноклеточная лимфома с расщепленным или нерасщепленным ядром.
Лимфомы высокой дифференциации:
• лимфобластная лимфома,
• диффузная мелкоклеточная недифференцированная лимфома Беркитта.
Лимфопролиферативные заболевания, протекающие с лейке- мизацией: в зависимости от распространенности процесса они подразделяются на следующие стадии:
стадия 0 — абсолютный лимфоцитоз без видимого увеличения лимфатических узлов;
стадия 1 — абсолютный лимфоцитоз и увеличение лимфатических узлов; стадия 2 — абсолютный лимфоцитоз и увеличение печени и (или) селезенки (с наличием лимфаденопатми или без нее); стадия 3 — абсолютный лимфоцитоз и анемия (НЬ меньше 110 г/л) с наличием или без увеличения лимфатических узлов, печени и (или)селезенки;
стадия 4 — абсолютный лимфоцитоз и тромбоцитопения с увеличением или без увеличения лимфатических узлов, печени и (или) селезенки.
По составу периферической крови различают:
— лейкемическийтип (количество лейкоцитов в периферической крови более 30x109/л),
— сублейкемический (количество лейкоцитов более 10х109/л) или,
— алейкемический тип (количество лейкоцитов в периферической крови менее 10х109/л).
По клинико-морфологическим формам выделяют:
• типичную медленно прогрессирующую форму (В-фенотип);
• опухолевую форму (В-фенотип);
• морфологическую атипичную (пролимфоцитарную, В-фенотип);
• волосатоклеточную, или ворсистоклеточную (В-фенотип);
• Т-клеточные лимфомы:
— синдром Сезари,
— грибовидный микоз.
Генерализованные плазмоцитомы (миеломная болезнь, множественная миелома) характеризуются пролифрацией В-лимфоцитов, дозревающих до плазматических клеток. Они могут быть различными по локализации:
— множественно-очаговая,
— диффузная,
— диффузно-очаговая.
По иммуннохимическим типам:
— G-миелома,
— А-миелома,
— D-миелома,
— Е-миелома,
— М-миелома,
— болезнь легких цепей (миелома Бенс-Джонса),
— несекретирующая миелома,
— диклоновая миелома.
V I
По течению:
— быстро прогрессирующая,
— медленно прогрессирующая.
По клиническим проявлениям различают варианты:
• с преобладанием костной патологии;
• с поражением внутренних органов;
• с миеломной нефропатией;
• с параамилоидозом;
• с синдромом недостаточности антител;
• с геморрагическим синдромом;
• с сенсорной периферической нефропатией;
• с синдромом повышенной вязкости, с гиперкальциемией. Макроглобулинемия Вальденстрема. Выделяются следующие
формы.
По стадиям:
— начальная,
— развернутая,
— кахектическая. По течению:
— медленно прогрессирующая,
— быстро прогрессирующая. По картине периферической крови:
— алейкемическая,
— сублейкемическая.
По клиническим проявлениям: малосимптомная;
гиперпластическая (с гепато- и/или спленомегалией, увеличением лимфатических узлов);
с гипервискозным синдромом (церебропатией, ретинопатией и кровоточивостью);
с геморрагическим синдромом; с анемическим синдромом; с периферической нейропатией.
Болезнь тяжелых цепей. По иммунохимическим типам: гамма-тип, дельта-тип, мю-тип, сигма-тип.
По клиническим проявлениям:
с увеличением периферических лимфатических узлов; с гепато- и спленомегалией;
абдоминальная форма (с инфильтрацией тонкой кишки и синдромом нарушенного всасывания);
легочная форма (с бронхопульмональными поражениями и медиас- тинальной лимфаденопатией); с остеолитическими поражениями. Злокачественные лимфомы. Лимфогранулематоз (болезнь Ходж- кина) разделяют на следующие виды. По гистологическим типам:
• лимфоидное преобладание,
• нодулярный склероз,
• смешанно-клеточный,
• лимфоидное истощение.
По распространенности процесса:
I стадия — поражение одной или двух смежных групп лимфатических уз
лов, расположенных по одну сторону диафрагмы, или наличие одного экстранодального инфильтрата (Е);
II стадия — поражение двух или более лимфатических узлов несмежных
групп, расположенных по одну сторону диафрагмы, или то же в сочетании с экстранодальным инфильтратом;
III стадия — поражение двух или более групп лимфатических узлов, рас
положенных по обе стороны диафрагмы, возможно наличие экстранодальных инфильтратов (IIIE) и поражение селезенки (NIC) или наличие и того, и другого (IIIEC);
IV стадия — поражение нелимфатических органов (костного мозга, легоч
ной паренхимы, плевры, печени, почек, пищеварительного тракта и т.д.).
Неходжкинские лимфомы подразделяют по гистологическому типу:
• нодулярные,
• диффузные.
По цитоморфологическим особенностям (Кильская классификация, К. Леннерт, 1988):
• лимфоцитарная (включая хронический лимфолейкоз) Т- и В-типа;
• лимфоплазмоцитарная (В-типа);
• лимфома Леннерта (Т-типа);
• плазмоцитарная (В-типа);
• центробластно-центроцитарная (В-типа);
• центроцитарная (В-типа);
• ангиоиммунобластная (Т-типа);
• лимфома Т-зоны;
• центробластная (В-типа):
• иммунобластная (В- иТ-типа);
• лимфома Беркитта (В-типа);
• лимфобластная (В- и Т-типа);
• плеоморфная (Т-типа),
• малт-лимфома.
Неклассифицируемые злокачественные лимфомы подразделяют по гистологии и распространенности процесса: нодулярный склероз с добавлением стадии V — лейкемизации или трансформации в лимфосар- комоклеточный или ретикулосаркомоклеточный лейкоз. По активности процесса: смешанно-клеточный. Фазы заболевания:
— активное течение,
— ремиссия.
Глава 20. Патология свертывания крови
Для понимания механизмов, способствующих нарушению системы свертывания крови, необходимо представить основные сведения о факторах, поддерживающих кровь в жидком состоянии у здорового человека, а также о механизмах, обеспечивающих целость сосудистой стенки и сохранение крови в сосудистом русле в случаях повреждения его структур.
20.1. Факторы, свертывающие кровь
и поддерживающие кровь в жидком состоянии
Нормальное формирование внутри сосудистого русла оптимального количества кровяных сгустков, обеспечивающих целость сосудов, а также растворение их избыточного образования осуществляют следующие компоненты крови и сосудистой стенки:
• прокоагулянты — белки крови, обеспечивающие ее свертывание, т.е. превращение из золя в гель;
• антикоагулянты — белки крови, ограничивающие процесс свертывания крови;
• тромбоциты — форменные элементы крови;
• фибринолитическая система крови, обеспечивающая растворение уже сформировавшегося фибрина;
• эндотелий сосудов.
Прокоагулянты. Известны компоненты крови, взаимодействие которых приводит к превращению жидкой крови в кровяной сгусток. Они пронумерованы Международным комитетом гемостаза и тромбоза и в большинстве своем обозначаются римскими цифрами, хотя сохраняют за собой и те названия, которые они получили от исследователей, их обнаруживших. К ним относятся следующие факторы:
I. Фибриноген
II. Протромбин
III. Тромбопластин
IV. Кальций
V. Проакцелерин
VI. Акцелерин
VII. Проконвертин
VIII. Антигемофилический глобулин А.
IX. Антигемофилический глобулин В.
X. Фактор Стюарт-Прауэр.
XI. Антигемофилический глобулин С. Предшественник тромбопласти- на плазмы.
XII. Фактор Хагемана.
XIII. Фибринстабилизирующий фактор.
• КВМВ. Кининоген высокой молекулярной массы. Кофактор контактной активации.
• ПК. Прекалликреин. Фактор Флетчера.
Из перечисленных факторов в крови определяются лишь 12 белков и кальций. До сих пор не удалось идентифицировать тромбопластин в свободном состоянии. Известно, что он располагается внутри многих клеток, выходя на поверхность лишь некоторой своей частью, обеспечивающей активацию процесса свертывания крови. Не удается также определить и акцелерин, который является активированной формой про- акцелерина и осуществляет свое действие лишь при соединении с фосфолипидной поверхностью. Физико-химические свойства прокоагу- лянтов представлены в таблице 20.1.
Таблица 20.1
Прокоагулянты — факторы свертывания крови
|
Антикоагулянты. К настоящему времени известны следующие факторы, регулирующие степень формирования фибрина: антитромбин III, или антитромбин, протеин С, протеин S и ингибитор пути тканевого фактора. Кроме них, имеются доказательства того, что процесс внутрисосу- дистого фибринообразования может сдерживаться также с помощью кофактора гепарина, называемого еще гепарин-кофактор II. Однако полагают, что он способен осуществлять определенное противотром- ботическое действие, контролируя избыток образования фибрина на поверхности эндотелия при взаимодействии с сульфированными муко- полисахаридами. Этот эффект рассматривается в качестве «противотром- ботического действия второго эшелона» (табл. 20.2).
Таблица 20.2
Антикоагулянтные факторы крови
|
Фибринолитическая система. В крови имеется определенная группа факторов, регулирующих интенсивность внутрисосудистого образования фибрина путем его растворения. Это происходит за счет действия протеолитического фермента плазмина (фибринолизина).
Плазмин образуется из постоянно присутствующего в плазме белка плазминогена (профибринолизина) под действием активаторов плазми- ногена — тканевого и мочевого. Последний получил свое название только потому, что впервые был обнаружен в моче, хотя этот фактор постоянно присутствует в плазме. Кроме описанных белков, в крови имеются компоненты, ограничивающие действие активного плазмина. К ним относятся антиплазмин и а2-макроглобулин, причем основная роль в инги- биции плазмина принадлежит антиплазмину, нейтрализующему до 80 % активного плазмина.
В крови имеются также субстанции, ограничивающие действие активаторов плазминогена. Они получили название ингибиторов активаторов плазминогена — ИАП-1 и ИАП-2, физиологическая функция последнего из них до сих пор точно не определена (табл. 20.3).
Таблица 20.3
Компоненты фибринолитической системы
|
| Компонент | Молекулярная | Место | Действие |
| масса | образования | ||
| а2-Антиплазмин | 67 ООО | Интактирует | |
| плазмин | |||
| Ингибитор | 50 000- | Гепатоциты, | Инактивирует |
| активатора | гликопротеин | эндотелий | и тканевый, |
| плазминогена-1 | и урокиназный | ||
| типы активаторов | |||
| плазминогена | |||
| Ингибитор | Гепатоцит, клетки | Подавляет актив | |
| активатора | ретикулоэндоте | ность активации | |
| плазминогена-2 | лиальной системы | ||
| плазмина |
Тромбоциты. Наименьшие безъядерные клеточные формирования крови размером от 3 до 5 мкм, содержатся в количестве 20x1012— 40х1012/л.
Тромбоциты способны видоизменяться, активироваться и подвергаться своеобразным превращениям. Изменяя структуру своей оболочки и активируя те или иные рецепторы, тромбоциты способны прилипать к дезэндотелизированной поверхности сосудистой стенки — к коллагену, микрофибриллам, а также к чужеродной поверхности, например к стеклу. Это явление получило название адгезии.
Адгезия к сосудистой поверхности осуществляется с помощью гли- копротеида тромбоцитарной оболочки GL-1b и фактора Виллебранда, белка плазмы крови, который является носителем прокоагулянта-факто- ра VIII.
Тромбоциты способны также объединяться друг с другом, что получило название реакции агрегации. Агрегация осуществляется с помощью других гликопротеидов тромбоцитарной оболочки — GL-II b и III а. В процессе активации тромбоцитов осуществляется выход из них в окружающую среду некоторых медиаторов, влияющих на механизмы гемокоагу- ляции. Этот феномен получил название реакции высвобождения.
Объединение тромбоцитов в единый конгломерат сопровождается также объединением сократительных актомиозиноподобных белков этих клеток, получивших название ретрактозимов. Они способствуют дальнейшей консолидации сгустка — его ретракции. Нарушение каждого из этих свойств тромбоцитов может приводить ктому или иному виду патологии — повышенной кровоточивости или повышенному тромбобразованию.
Эндотелий сосудов. Выяснено, что эндотелиальные клетки сосудов способны активно участвовать в процессе формирования внутрисо- судистого фибриново-тромбоцитарного сгустка и его растворения. Это осуществляется за счет выработки эндотелием таких простагландинов, как тромбоксан Тх-А2 и простациклин Pg-I2, регулирующихтромбоцитар- ные процессы адгезии и агрегации, а также тканевого активатора плаз- миногена, мочевого активатора плазминогена, фактора Виллебранда, тромбомодулина и некоторых других субстанций, участвующих в процессах гемокоагуляции. Представленные данные не могут быть игнорируемы при оценке причин нарушений гемокоагуляции как при формировании тромбозов, так и при повышении кровоточивости.
Современные представления о физиологической гемокоагуляции. Обнаружение в крови каждого человека таких маркеров фибрино- образования, как Д-димер, растворимые комплексы фибрин-мономера, фибринопептид-А, (3-тромбоглобулин, 4-й фактор тромбоцитов и некоторые другие, заставляет сделать вывод о том, что процесс гемокоагуляции происходит перманентно. Каков физиологический смысл этого явления? Существует предположение о том, что непрерывное функционирование гемокоагуляции необходимо для репарации постоянно возникающих дефектов эндотелия. Есть и другое мнение: непрерывное фиб- ринообразование необходимо для того, чтобы обеспечивать клетки важным для их существования «пластическим белком». Так или иначе, образование фибрина и тромбоцитарно-фибринового сгустка постоянно имеет место, и осуществляется это следующим образом.
После нарушения целости эндотелиальной поверхности обнажившиеся микрофибриллы или коллаген осуществляют взаимодействие с фактором Виллебранда. Последний контактируете рецептором тромбоцитов lb и обеспечивает адгезию тромбоцитов в этом месте. В процессе адгезии тромбоцитов происходит активация иных рецепторов на их поверхности. Активированные гликопротеиды lib—Ша обеспечивают объединение тромбоцитов друг с другом. В этом им способствуют молекулы фибриногена. Одновременно происходит высвобождение из тромбоци- тарных гранул таких биологически активных субстанций, как АДФ, серо- тонин, создается возможность для экспозиции фосфолипидных участков, обеспечивающих взаимодействие на них факторов гемокоагуляции и формирование тромбина, а за ним и фибрина. Механизм формирования фибриновой сети хорошо известен.
1. Фактор VII контактирует с тканевым фактором и образует с ним комплекс, в котором фактор VII уже становится активным.
2. Комплекс обеспечивает активацию факторов IX и X.
3. Фактор Ха, взаимодействуя с фактором Va и протромбином на фос- фолипидной поверхности, приводит к образованию тромбина.
4. Тромбин в небольших количествах способствует активации факторов VIII, V, XI, превращая их в Villa, Va, Xla.
5. Происходит также интенсивная активация тромбоцитов, на поверхности которых осуществляется взаимодействие активного фактора IXa с активными факторами Villa и Va; это приводит к образованию больших количеств тромбина, способного образовывать фибрин из фибриногена путем отщепления фибринопептидов А и В через фибрин-мономеры, объединяющиеся затем в полимеры фибрина и укрепляющиеся фибринстабилизирующим фактором, также активируемым тромбином.
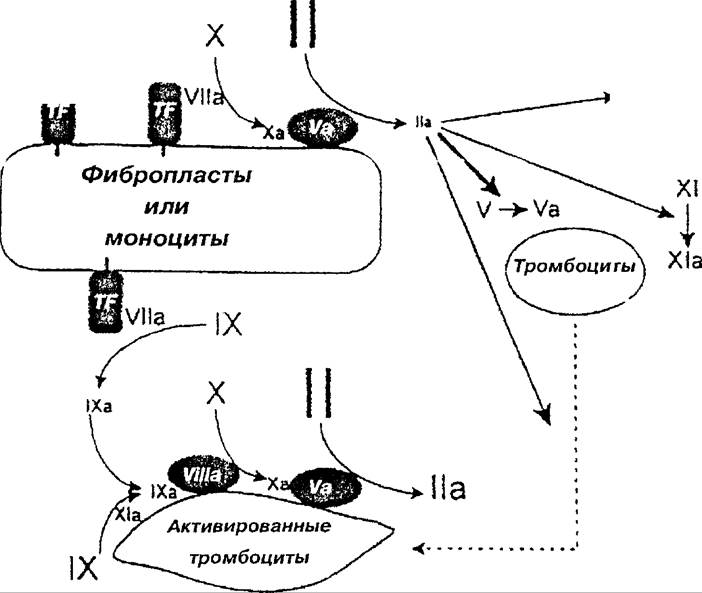
|
| TFPI XagBViia |
| VIll/vWF i Villa + free vWF |
| Рис. 20.1. Схема реакций, следующих за активацией факторов X и IX комплексом TF-Vlia. Она отражает комбинированное влияние факторов на конечное образование тромбина. Необходимо отметить, что малое количество тромбина, формируемое комплексом TF-Vlla, достаточно для активации тромбоцитов, кофакторов, а также фактора XI. Затем фактор Xla способствует большему образованию фактора IXa из фактора IX, усиливая тем самым гененерацию тромбина TFPI — ингибитор пути тканевого фактора; VWF — фактор Виллебранда; TF — тканевый фактор; Ха, Vila, Xla и пр. — активированные факторы свертывания |
Дата добавления: 2015-02-05; просмотров: 975;