Механизмы химических реакций
Для аренов характерны три типа реакций:
1) электрофильное замещение SEAr (разрушение связи С-Н);
2) реакции присоединения (разрушение p-связи);
3) реакции с разрушением бензольного кольца.
Электрофильное замещение в аренах (SEAr)
Реакции электрофильного замещения протекают по общей схеме через образование π- и σ-комплексов
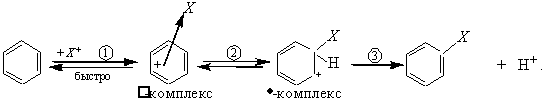
Как следует из представленной схемы, ароматическое замещение SEAr протекает по механизму присоединения – отщепления. За присоединением электрофильного агента Х+ к ароматическому субстрату с образованием σ- комплекса следует отщепление протона с образованием продукта реакции.
Реакции электрофильного замещения в аренах, как правило, следуют по кинетическому уравнению второго порядка (v = k2[X+][Ar-H]).
Рассмотрим постадийное протекание процесса.
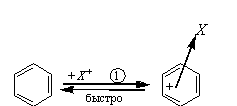
Стадия 1. Образование π- комплексов
 .
.
π–Комплексы –координационные соединения, в которых донором электронов является ароматическое соединение, имеющее легко поляризуемые π–электроны. π–комплексы – не классические химические соединения, в которых электрофильная частица связывается с ковалентной связью с каким-либо ковалентным атомом реагирующего вещества. Большинство π–комплексов легко разлагается при нагревании или при воздействии воды.
Способность к образованию π–комплексов у аренов возрастает в ряду:
C6Н6< C6Н5СН3< п - СН3–С6Н4–СН3 ~ п - СН3–О–С6Н4СН3<
<м - СН3–С6Н4-СН3< 1,3,5 (СН3)3С6Н3
Чем большей π–электронной плотностью обладает соединение, тем с большей легкостью оно образует π–комплексы.
Стадия 2. Образование σ- комплексов
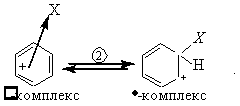
σ-Комплексы – это катионы, при образовании которых реагент Х+ образует ковалентную связь с одним из атомов углерода за счет 2-х π-электронов бензольного ядра, при этом этот С-атом переходит из sp2-состояния в sp3-гибридизацию, в которой все четыре его валентности находятся под углом ~1090. Симметрия бензольного ядра нарушается. Группа Х и атом водорода оказываются в плоскости перпендикулярной плоскости бензольного ядра.

Устойчивость σ–комплексов возрастает с увеличением основности бензольного ядра
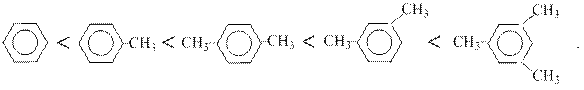
Эта стадия является самой медленной стадией всей реакции и называется лимитирующей.
Стадия 3. Отщепление протона от σ – комплекса
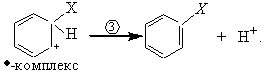
В последней стадии происходит отщепление от σ–комплекса протона и восстановление 6π–электронного облака (ароматической структуры). Этот процесс протекает с выигрышем энергии ~ 42 кДж/моль. Во многих реакциях отрыву протона на заключительной стадии способствует соответствующее основание, присутствующее в растворе.
По рассмотренному механизму в аренах протекают следующие реакции.
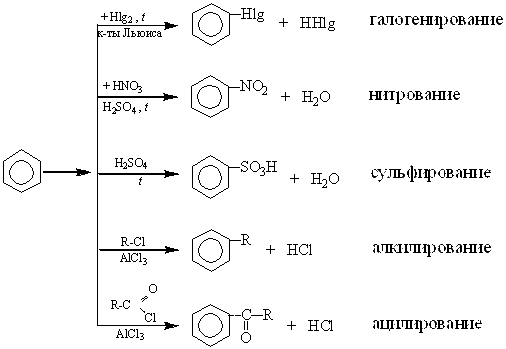
Однако предложенную схему не следует рассматривать как абсолютно доказанную и универсальную. В различных процессах на ход реакции оказывает влияние:
Ø структура субстрата;
Ø химическая активность реагента;
Ø условия проведения процесса;
Ø характер, активность катализатора и другие факторы, что может приводить к отклонению в частных случаях от предложенной схемы процесса.
Рассмотрим некоторые примеры электрофильного замещения в бензоле.
Пример 1. Бромирование бензола
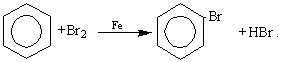
Молекулярный бром является слишком слабым электрофильным агентом и, в отсутствии катализатора, не реагирует с бензолом.
Чаще всего реакцию бромирования бензола осуществляют в присутствии бромида железа (III), играющего роль кислоты Льюиса, последний получают в реакционной массе непосредственным взаимодействием брома с железом
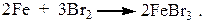
Стадия 1. Образование электрофильного реагента Е+.
Молекула брома активируется по схеме кислотно–основной реакции с кислотой Льюиса.
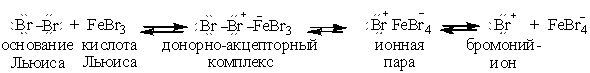
Стадия 2.Образование π – комплекса 1.
Свободный бромониевый ион или ион в составе ионной пары является активным электрофильным агентом, способным реагировать с бензолом; при этом сначала образуется π-комплекс 1
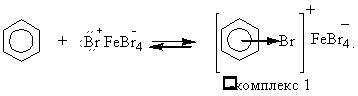
Роль электрофильного агента на этой стадии может выполнить и донорно–акцепторный комплекс 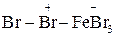 .
.
Стадия 3. Перегруппировка π-комплекса 1 и образование σ- комплекса, или аренониевого иона.
Это наиболее медленная стадия всей реакции
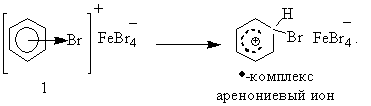
Стадия 4. Перегруппировка σ-комплекса в π-комплекс 2 продукта замещения. Протон отщепляется от атома углерода, у которого проходит замещение; в цикле вновь формируется ароматический секстет электронов – наблюдается реароматизация
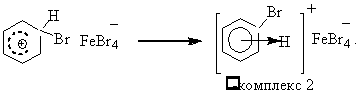
Стадия 5. Диссоциация π-комплекса 2 с образованием продукта замещения

Механизм электрофильного бромирования бензола иллюстрируется энергетической диаграммой реакции, показанной на рис.11.
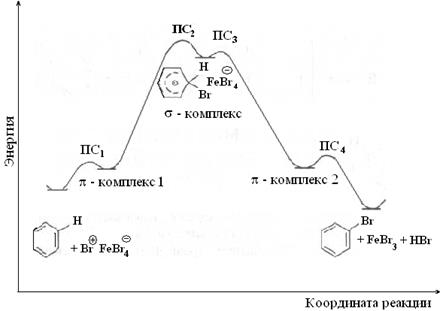
Рис. 11. Энергетическая диаграмма реакции
электрофильного бромирования бензола;
ПС – переходное состояние.
Стадии 2 и 5, включающие π–комплексы исходного арена и продукта замещения, в схемах механизма электрофильного ароматического замещения часто опускают. При таком подходе собственно электрофильное ароматическое замещение включает лишь три стадии.
Стадия 1' – образование электрофильного агента.
Стадия 2' – образование σ-комплекса, минуя π-комплекс 1.
Стадия 3' – распад σ-комплекса с образованием продукта замещения, минуя π-Комплекс 2.
Пример 2. Нитрование аренов
Нитрование заключается в замещении атома водорода бензольного кольца на нитрогруппу NO2. Бензол реагирует с концентрированной азотной кислотой медленно даже при нагревании. Поэтому нитрование чаще всего проводят действием более активного нитрующего агента – нитрующей смеси – смеси концентрированных азотной и серной кислот. Нитрование аренов нитрующей смесью является основным способом получения нитросоединений ароматического ряда

Нитрование бензола нитрующей смесью проводят при 45–500С. Поскольку реакция нитрования необратима, азотную кислоту применяют в минимальном избытке (5–10%), добиваясь практически полного превращения бензола.
Серная кислота в составе нитрующей смеси необходима для повышения концентрации электрофильного агента – нитроний-иона NO2+.
Стадия 1. Образование электрофильного агента.
Действующим электрофильным агентом при нитровании является ион нитрония 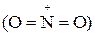 , который потенциально имеется в целом роде соединений.
, который потенциально имеется в целом роде соединений.
 Например: HO _ NO2, O2N _ O _ NO2, и др.
Например: HO _ NO2, O2N _ O _ NO2, и др.
Их склонность к образованию иона нитрония увеличивается с повышением электроотрицательности заместителя, связанного с нитрогруппой.
Гидроксильная группа как таковая отщепляться не может, поэтому ион нитрония из азотной кислоты образуется только в кислой среде
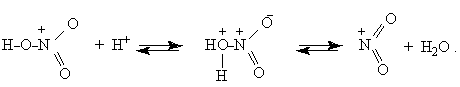
В простейшем случае азотной кислота может протонироваться сама («самопротонизация»)

Однако равновесие смещено в левую сторону, поэтому азотная кислота нитрует слабо.
При добавлении концентрированной серной кислоты концентрация 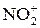 - катиона сильно повышается
- катиона сильно повышается

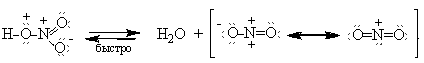
Нитрующее действие смеси азотной и серной кислоты (нитрующей смеси) намного сильнее, чем одной азотной кислоты. Дальнейшее повышение реакционной способности можно достигнуть, применяя дымящую азотную кислоту и олеум.
Стадия 2. Образование σ-комплекса

Стадия 3. Выброс протона с образованием продукта замещения
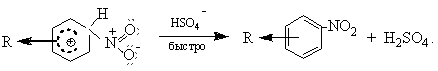
На практике необходимо согласовывать активность нитрующего средства с реакционной способностью ароматического ядра.
Так, например, фенолы и простые эфиры фенолов нитруются уже разбавленной азотной кислотой, нитрование же бензальдегида, бензойной кислоты, нитробензола и т.д. требует смеси дымящей азотной кислоты с серной.
м-Динитробензол с трудом нитруется даже смесью дымящей азотной и серной кислот (5 сут, 1100С; выход 45 %).
При нитровании наиболее часто побочной реакцией является окисление. Ему благоприятствует повышение температуры реакции. Процесс окисления определяют по выделению окислов азота. Альдегиды, алкиларил – кетоны и в меньшей степени алкилбензолы при нитровании также подвержены окислению.
Пример 3. Алкилирование аренов
В качестве алкилирующих средств могут быть использованы R-HIg, ROH, R-CH=CH2 в присутствии соответствующих катализаторов (в частности AICI3, AIBr3, H2SO4).
Катализаторы генерируют (образуют) электрофильную частицу – карбкатион
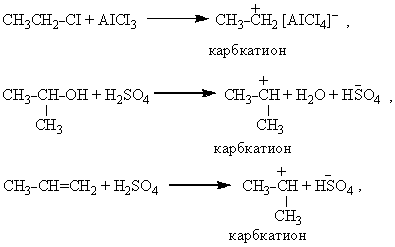
который далее по известной схеме участвует в реакции.
Реакции алкилирования имеют три серьезных ограничения:
1) реакцию трудно остановить на стадии моноалкилирования, т.е. она протекает дальше, с образованием полиалкилбензолов; для подавления полиалкилирования обычно используют избыток арена;
2) если в арене только электроакцепторные заместители (например, -NO2), то реакцию алкилирования не удается осуществить;
3) реакция алкилирования сопровождается перегруппировкой алкильного радикала.
Перегруппировка алкильного радикала в наиболее устойчивый является характерным свойством карбкатионов
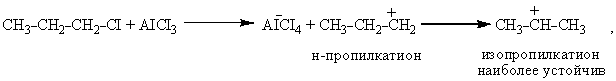
поэтому
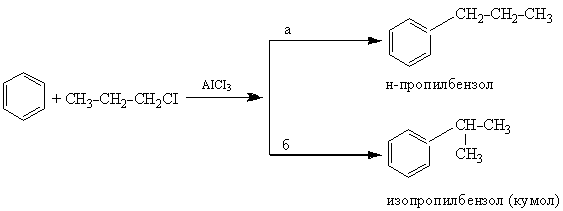
соотношение а:б = 1:2.
Пример 4. Ацилирование
Эта реакция сходна с алкилированием по Фриделю – Крафтсу.
В качестве ацилирующих агентов используют галогенангидриды

 или ангидриды карбоновых кислот.
или ангидриды карбоновых кислот.
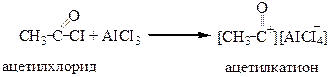
Ацетилкатион так активен, что катализатор Фриделя – Крафтса остается связанным с ним, поэтому по меньшей мере необходимо использовать в реакции эквимолекулярное количество катализатора:
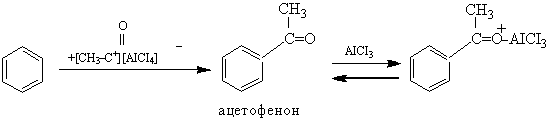
Ангидриды требуют более двух молей катализатора:
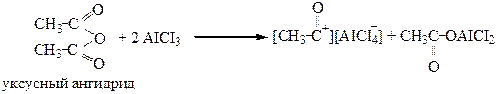
Дальнейшее ацилирование вследствие дезактивации ароматического ядра практически невозможно.
Разложение комплекса по окончании реакции ацилирования осуществляют соляной кислотой со льдом.
Правила ориентации
Реакции замещения водорода в бензоле идут при любом атоме углерода одинаково, так как молекула бензола симметрична. Однако если в бензоле уже имеется заместитель, то положения, остающиеся свободными для реакций электрофильного замещения, становятся неравноценными.
Закономерности, определяющие направления реакций замещения в бензольном ядре, называются правилами ориентации.
–Активирующая группа – заместитель, который делает кольцо бензола более реакционно способным в реакциях электрофильного замещения по сравнению с незамещенным бензолом.
–Дезактивирующая группа – заместитель, который делает бензольное кольцо менее реакционноспособным в реакциях электрофильного замещения по сравнению с незамещенным бензолом.
– о-, п-ориентант – заместитель, направляющий атаку электрофила преимущественно в о- или п-положение бензольного кольца.
– м-ориентант – заместитель, направляющий атаку электрофила преимущественно в м-положение бензольного кольца.
В общем случае электрофильное замещение в монозамещенном бензоле может протекать в трех направлениях
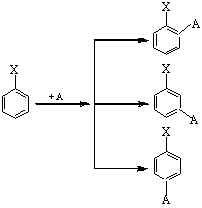
Реакционная способность атомов углерода в этом случае определяется тремя факторами:
1) природой уже имеющегося заместителя;
2) природой действующего агента;
3) условиями проведения реакции.
По своему влиянию на ориентацию в этих реакциях все заместители делятся на две группы: заместители первого рода (орто-, пара-ориентанты) и заместители второго рода (мета-ориентанты).
Дата добавления: 2016-02-16; просмотров: 14101;