Гидроксиды, соли и комплексные соединения металлов 11 группы.
Химия кислородных соединений металлов 11 группы определяется степенью окисления элемента, то есть степенью заполнения d-подуровня, а также размером иона, его окислительно-восстановительными свойствами. Химия водных растворов устйочивых на воздухе в отсутствие восстановителей и анионов-комплексообразователей представлена исключительно соединениями меди(II), серебра(I) и золота(III).
Степень окисления +1.
В степени окисления +1 элементы группы меди имеют электронную конфигурацию d10, что соответствует полностью заполненному d-уровню. Таким образом, соединения меди(I), серебра(I) и золота(I) не имеют энергетического выигрыша связанного со стабилизацией в кристаллическом поле в независомости от геометрии частицы. В этом состоит существенное отличие этих соединений от от аналогичных их соединений переходных металлов предшествующих групп. Геометрия координационного окружения ионов M+ определяется исключительно взаимным отталкиванием лигандов, что обусловливает низкие координационные числа – два, три и четыре, которым соответствуют линейная, треугольная и тетраэдрическая форма ионов и молекул. Невозможность d-d переходов обусловливает отсутствие окраски, которая может быть вызвана лишь переносом заряда с аниона на катион (Cu2O, Cu2S, Ag2S).
Степень окисления +1 является промежуточной для всех трех элементов, что позволяет предположить возможность диспропорционирования. Оно наиболее типично для соединений золота(I) и меди(I) и совсем нехарактерно для серебра(I), для которого данная степень окисления наиболее устойчива в растворах.
Соединения меди(I) образуются при восстановлении меди(II) гидразином, хлоридом олова(II), сернистым газом, иодид- и цианид-ионами. Равновесие диспропорционирования
2Cu+  Cu2+ + Cu
Cu2+ + Cu
в газовой фазе сильно смещено влево – в сторону образования катионов меди(I). В кристаллах и в растворе на значение константы существенное влияние оказывают молекулы растворителя и анионы. Так, в водном растворе в отсутствие комплексообразователей равновесие смещается вправо благодаря тому, что энергия сольватации ионов Cu2+ (2100 кДж/моль) более чем в три раза превышает энергию сольватации ионов Cu+ (580 кДж/моль) – это объясняется существенно разницей в ионных радиусах и зарядах ионов. В менее донорных растворителях, например, ацетоне, преобладают ионы меди(I). Смещению равновесия влево способствует также ацетонитрил, образующие прочные сольваты с ионом меди(I). Стабилизацию степени окисления +1 вызывают лиганды, способные образовывать линейные или треугольные комплексы, такие как галогенид-ионы, молекулы аммиака.
Наиболее устойчивы комплексы, в которых ионы меди(I) связаны в бесконечные цепи или плоские слои линейными полидентатными анионами. Примером является дицианокупрат(I) [Cu(CN)2]–, построенный из бесконечных плоских зигзагообразных цепей с плоско-треугольными фрагментами Cu(CN)3, связанных мостиковыми цианидными группами (Рис. 7.11. Строение [Cu(CN)2]– в K[Cu(CN)2], ПРОСТАВИТЬ РАССТОЯНИЯ: Cu-C(цепь) 0.1951 нм, Сu-C(терм) 0.1919 нм, Cu-N 0.2047 нм). Расположение атомов меди и цианидных групп в одной плоскости указывает на частичное перекрывание заполненных d-орбиталей атомов меди и вакантных разрыхляющих π*-орбиталей цианид-ионов.
Ионы меди(I) благодаря тяготению к линейной геометрии оказываются неустойчивыми в окружении хелатирующих лигандов, не способных выполнять роль линейных мостиков, таких как этилендиамин или ацетилацетон (Табл. 7.5.). В то же время соединения меди(II) с этими лигандами благодаря хелатному эффекту необычайно устойчивы. Именно поэтому, они смещают равновесие диспропорционирования вправо. Так, при добавлении к соли меди(I) этилендиамна образуется синий раствор комплекса меди(II) и выпадает осадок меди:
2CuCl + 2en = [Cuen2]2+ + 2Cl– + Cu¯
Таблица 7.5. Равновесие диспропорционирования меди(I) в присутствии различных лигандов и константы устойсивости комплексов
| Лиганд | Контанта равовесия
2Cu(I) Cu(II) + Cu
в растворе лиганда
| Десятичный логарифм константы устойчивости K1,2 комплекса | ||
| Cu(I) | Cu(II) | |||
| H2O | 106 | |||
| NH3 | 2×10–2 | 10.86 | 7.33 | |
| en* | 105 | 10.79 | 20.13 | |
| Cl– | ПОСЧИТАТЬ | 5.35 | –0.57 | |
| Br– | ПОСЧИТАТЬ | 5.92 | –0.30 | |
| I– | 8.85 | ? | ||
| CN– | 24.0 | ? | ||
| SCN– | ПОСЧИТАТЬ | 9.90** | 5.19** | |
* для пентаметилэтиледимина, не способного образовывать хелатные цикл по стерическим причинам константа диспропорционирования равна 3×10–2
** K1-3
Таким образом, медь(I) может быть стабилизирована в комплексах с иодидными, цианидными и тиоцианатными лигандами, а также в форме малорастворимых в воде оксида Cu2O, сульфида Cu2S, галогенидов, особенно иодида CuI. В то же время хелатирующие лиганды и анионы кислородсодержащих кислот отдают предпочтение ионам меди(II).
Ионы меди(I) существуют в водном растворе лишь в концентрации менее 0,01 моль/л, которая отвечает равновесию диспропорционирования. Более крепкие растворы могут быть получены восстановлением солей меди(II) ионами ванадия(II) или хрома(II), однако в течение нескольких часов концентрация ионов Cu+ понижается до сантимолярной вследствие диспропорционирования.
Попытки осаждения гидроксида меди(I) как указывалось ранее приводят к образованию мелкодисперсной формы оксида Cu2O. Гипотетическое основание CuOH слабо амфотерно, с заметным преобладанием основных свойств.
Купраты(I) получают сплавлением оксида меди(I) и оксида или азида щелочного металла. В некоторых случаях используют смесь оксидов меди(I) и (II):
450°C, 50 ч
2CuO + Cu2O + 2CsN3 ¾¾¾¾¾® 2CsCu3O2 + 3N2.
Купраты состава MCuO содержат плоские циклические анионы Cu4O44– (Рис. 7.12. Строение купратов меди(I): (а) анион Cu4O44– в структуре KCuO, (б) фрагмент слоя [Cu3O2]nn– в структуре CsCu3O2), соли M3CuO2 – линейные анионы [O-Cu-O]3–, M3Cu5O4 – спирали из цепей -O-Cu-O-, MCu3O2 – слои из конденсированных циклов Cu6O6 (M. Sofin, E.M. Peters, M. Jansen, Z. Anorg. Allg.Chem., 2003, 629, 2435). В водных растворах они диспропорционируют:
2KCuO + 2H2O =Cu(OH)2 + Cu + 2KOH.
Из солей оксокислот наиболее известен бесцветный сульфат Cu2SO4, который удобно синтезировать взаимодействием меди с безводной серной кислотой или нагреванием оксида меди(I) с диметилсульфатом
Cu2O + (CH3)2SO4 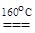 Cu2SO4 + (CH3)2O.
Cu2SO4 + (CH3)2O.
Выделяющийся сульфат меди (I) устойчив лишь в сухой атмосфере. При нагревании или под действием воды диспропорционирует
Cu2SO4 = Cu¯ + CuSO4,
а кислородом воздуха окисляется до смеси оксида и сульфата меди(II).
Перламутрово-белый кристаллический осадок сульфита меди(I), назывемый солью Этара, выпадает при пропускании сернистого газа через кипящий раствор ацетата меди(II):
2Cu(CH3COO)2 + 2SO2 + 3H2O = Cu2SO3¯ + 4CH3COOH + H2SO4
При сливании растворов сульфата меди(II) и сульфита натрия выпадает желто-оранжевый осадок соли Шевреля Cu3(SO3)2×2H2O, содержащий медь сразу в двух степенях окисления: Cu+2(Cu+1SO3)2×2H2O. Сухая соль устойчива на воздухе, в влажная постепенно окисляется, превращаясь в оксосульфат меди(II). При нагревании без доступа воздуха она превращается в смесь оксида меди(I) и сульфата меди(II). Известны также и комплексные сульфиты меди(I), например, Na3[Cu(SO3)2] (И.А. Леенсон, Занимательная химия, часть1, М.,Дрофа, 1996, с. 126).
Нитрат меди(I) не описан, однако может быть выделен в виде комплекса с тиомочевиной Cu4tu9(NO3)4. Смешанный фосфат меди(I, II) Cu+1Cu+2PO4 получен по реакции между ДОПИСАТЬ ПО ССЫЛКЕ!!! + строение (Etheredge K.M.S., Hwu S.-J., Inorg. Chem., 1995, 34, 5013).
Ацетат меди(I) CuCH3COO выделяется при действии уксусной кислотой на раствор, образующийся при растворении оксида меди(I) в насыщенном растворе ацетата аммония. Синтез проводят в инертной атмосфере. Другой способ получения заключается в сопропорционировании ацетата меди(II) и меди в пиридине или ацетнитриле, смещающих вправо равновесие Cu(II) + Cu 2Cu(I) благодаря сольватированию ионов меди(I). Вещество имеет полимерное строение с мотиковыми η2-ацетатными группами:
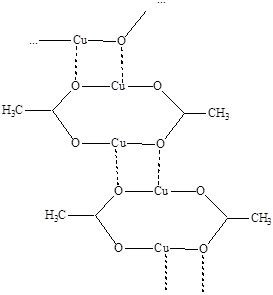
В воде оно полностью гидролизуется, превращаясь в исходный оксид меди(I), а под действием раствора серной кислоты диспропорцонирует на сульфат меди(II) и медь.
Красно-бурый порошок ацетиленида меди(I) Cu2C2 выпадает в осадок при пропускании ацетилена через аммиачный раствор хлорида меди или образуется при взаимодействии иодида меди с ацетиленидом калия. При охлаждении смеси сухим льдом (-78°C) может быть получен оранжевая соль CuCºCH, которая при -45°C разлагается на Cu2C2 и ацетилен. В соляной кислоте ацетиленид меди(I) растворвется с образованием хлоридного комлпекса H[CuCl2] и выделением ацетилена. При стряхивании или сотрясении сухая соль взрывает.
При действии азида калия на раствор сульфата меди(II) в присутствии восстановителя (сульфита) может быть выделен белый осадок азида CuN3, сотоящий из зигзагообразных цепей -Cu-N=N=N-Cu-.
Удобными исходными соединениями меди(I) являются ярко-красный оксид Cu2O и бесцветный хлорид CuCl, при растворении которых в концентрированной соляной кислоте образуется бесцветный раствор дихлорокупрата, в котором присутствуют линейные ионы [Cl-Cu-Cl]–:
Cu2O + 2HCl = H[CuCl2] + H2O
CuCl + HCl = H[CuCl2]
При разбавлении водой комплекс разрушается, и хлорид меди(I) выделяется в виде белого осадка.
При увеличении концентрации хлорид-ионов образуются комплексы [CuCl3]2–имеющие геометрию правильного треугольника, а тетрахлорокупрат-ионы [CuCl4]3–даже при сильном избытке хлорид-ионов присутствуют лишь в незначительном количестве. В структуре твердых хлорокупратов(I) присутствуют цепи из тетраэдров CuCl4 с общими ребрами ([Cu(NH3)4]Cu2Cl4) или вершинами (K2CuCl3, CsCu2Cl3) (Рис. 7.13. Фазовая диаграмма системы KCl-CuCl). Бромокупраты(I) построены аналогично, а многочисленные иодокупраты имеют еще более сложное строение (Cu2I42–, Cu3I4–, Cu8I135–, Cu36I5620– и др.), будучи образованными из тетраэдрических CuI4 и пирамидальных CuI3 блоков, связанных атомами иода (H. Hartl et al, Z. Anorg. Allg. Chem., 1988, 563, 96; 1991, 598, 151; Angew. Chem., Intern. Ed. Engl., 1986, 25, 569; 1994, 33, 1841).
При действии на исходные вещества или на раствор хлорокупрата(I) избытком аммиака может быть получен аммиакат, также не имеющий окраски
CuCl + 3NH3 + H2O = [Cu(NH3)2]OH + NH4Cl
Образование голубого или синего раствора свидетельствует об окислении комплекса до соединения меди(II):
Устойчивость комплексов меди(I) напрямую зависит от степени перекрывания заполненных d-орбиталей металла и вакантных p*-орбиталей лиганда. Именно благодаря этому π-дативному взаимодействию константы устойчивости галогенидных комплексов возрастают в ряду F < Cl < Br < I, соответствующему последовательному понижению энергии p*-орбиталей галогена. Напомним, что в комплексах переходных металлов с неполностью заселенным d-подуровнем, где определяющим фактором является величина расщепления кристаллическим полем, наблюдается обратная зависимость: F >Cl > Br > I. У меди, серебра и золота в стпени окисления +1 наиболее устойчивыми оказываются комплексы с π-акцепторными лигандами такими как цианид, тиоцианат и карбонил.
Смешение растворов сульфата меди(II) и цианида калия сначала приводит к образованию раствора цианидного комплекса меди(II), который уже при слабом нагревании разлагается на дициан и цианид меди(I), выделяющийся из раствора в виде белого осадка.
2CuSO4 + 4KCN = 2CuCN¯ + (CN)2
Цианид меди(I) хорошо растворим в водных растворах цианидов с образованием различных цианокупратов(I): ион [Cu(CN)2]–, имеет линейное строение, [Cu(CN)3]2–представляет собой плоский треугольник, [Cu(CN)4]3– – правильный тетраэдр. (Сноска: Гидротермальным синтезом получены и более сложные комплексы, например, Rb[Cu2(CN)3], в струткуре которого имеются кольца (CuCN)6 , T. Pretsch, I. Brudgam, H. Hartl, Z. Anorg. Allg. Chem., 2004, 630, 353).
При пропускании угарного газа через солянокислый раствор хлорида меди(I) образуется карбонильный комплекс Cu(CO)Cl, который может быть выделен в твердом виде при проведении реакции в органическом растворителе. Длительное время предполагалось, что вещество представляет собой димер с мостиковыми атмоами хлора. Возможно, некоторая доля димерных молекул присутствует в растворе наряду с частицами CuCOCl и [Cu(CO)(H2O)2]+. Изучение кристаллической структуры вещества однозначно доказало его полимерное строение. Каждый атом меди находится в центре тетраэдра, образованного одной молекулой СО и тремя мостиковыми атомами хлора (Рис. 7.14. Фрагмент слоя в структуре CuCOCl) (см. также том 2, стр. 125). Твердый карбонилхлорид меди(I) устойчив только под давлением угарного газа, а в открытом сосуде при хранении разлагается на CuCl и СО. Нагревание раствора этого соединения также приводит к выделению угарного газа. Солянокислый раствор хлорида меди(I), таким образом, является удобным реагентом для обратимого поглощения угарного газа.
Для серебра степень окисления +1 наиболее устойчива в водных растворах, поэтому известно большое количество солей с различными анионами. Большинство из них малорастворимо в воде – исключение составляют нитрат, хлорат, перхлорат и фторид. Особенно высока растворимость нитрата серебра: в 100 г воды при 20°C растворяется 222,5 г соли, а при 100°C – 770 г! Соли серебра получают растворением металла или оксида в кислотах, а малорастворимые в воде – с помощью реакций обмена
AgNO3 + NaΝO2 = AgΝO2¯ + NaNO3.
Все соли серебра чувствительны к свету, под действием которого они чернеют, восстанавливаясь до серебра.
Нитрат серебра AgNO3, или ляпис – бесцветное кристаллическое вещество (т.пл. 212оС), хорошо растворимое не только в в воде (Рис. 7.15. Растворимость нитрата серебра в воде), но и в полярных органических растворителях – этиловом спирте (2,12 г в 100 г этанола при 20°C), ацетонитриле, пиридине, ацетоне. При нагревании он разлагается, превращаясь в металлическое серебро:
350оС
2AgNO3 = 2Ag + 2NO2 + O2.
Работать с этим веществом надо осторожно, так как при попадании на кожу соли серебра вызывают ожоги, которые становятся заметными через несколько часов после их возникновения: в месте контакта с ионами серебра кожа темнеет, а затем отмирает. Именно на этом основано использование ляписа в медицине для прижиганий и удаления бородавок. Нитрат серебра также применяют для изготовления серебряных зеркал, как реагент в аналитической химии и исходное вещество для синтеза других соединений серебра.
Благодаря сильным окислительным свойствам иона Ag+ (E°(Ag+/Ag) = 0.799B) соединения серебра под действием восстановителей (цинк, муравьиная кислота, сульфат железа(II)) чернеют, а при нагревании разлагаются, окисляя анион:
500°C
AgClO4 = AgCl + 2O2
270°C
2AgClO3 = 2AgCl + 3O2
1000°C
Ag2SO4 = 2Ag + SO2 + O2.
Сульфат серебра Ag2SO4 (т. пл. 660°C) получают действием на металл горячей концентрированной серной кислоты. Он малорастворим в воде, но переходит в раствор под действием серной кислоты, образуя кислые соли AgHSO4, Ag(H3O)(HSO4)2, Ag2(HSO4)2(H2SO4), которые разлагаются водой (Stiewe A., Kemnitz E., Troyanov S., Z. Anorg. Allg. Chem., 1999, 625, 329).
Сульфит натрия вызывает осаждение белой средней соли Ag2SO3, растворимой в избытке реагента с образованием комплексных ионов [Ag(SO3)(H2O)2]–, [Ag(SO3)2]3–. При кипячении водного раствора осадок сульфита чернеет вследствие разложения на сульфат, серебро и сернистый газ.
Нитрит серебра AgNO2 на свету чернеет, при длительном кипячении раствора разлагается на нитрат, серебро и оксид азота(II), а при нагревании твердой соли до 140°C – на серебро и NO2.
Кислый фосфат натрия Na2HPO4 осаждает из нейтральных растворов солей серебра желтый осадок среднего фосфата Ag3PO4, растворимый в кислотах и аммиаке. Под действием метафосфата натрия выпадает белый осадок AgPO3. Известны также циклометафосфаты Ag3P3O9 и Ag6P6O18, а также кислые соли Ag2H2P2O7, Ag2HPO4.
Описано большое количество силикатов серебра: Ag2SiO3, Ag6Si2O7, Ag10Si4O13, Ag18(SiO4)2(Si4O13) (M. Jansen et al, Z. Anorg. Allg. Chem., 1991, 601, 5; 1996, 62, 486), а также смешанные соли Ag6(SiO4)(SO4) и Ag9(SiO4)2NO3 (Keller H.L., Mueller-Buschnaum H., Z. Anorg. Allg. Chem., 1974, 408, 205). Метасиликат серебра Ag2SiO3 построен из цепей. Образованных кремний-кислородными тетраэдрами, между которыми расположены ионы Ag+ (Рис. 7.16. Строение Ag2SiO3).
Ацетат серебра CH3COOAg кристаллизуется в виде плоских игл из раствора, полученного действием уксусной кислоты на карбонат или оксид серебра.
Белые осадки азида AgN3, цианида AgCN и роданида AgSCN серебра получают по обменным реакциям. Фульминат серебра AgCNO образуется при нагревании до начала кипения раствора нитрата серебра в азотной кислоте и этиловом спирте. Вещество существует в ввиде двух полиморфных модификаций, в каждой из которых атом серебра координирован атомами углерода двух фульминат-ионов, сединяющих атомы серебра в цепи или циклические гексамеры. При нагревании или ударе азид и фульминат серебра разлагаются со взрывом: 2AgCNO = 2Ag + 2CO + N2. У фульмината серебра есть изомер – цианат серебра AgNCO – полимер, в котором атомы серебра координированы атомами азота лиганда. Связь Ag-N имеет большую энергию, чем Ag-C, поэтому цианат выдерживает нагревание до 335°C и не является взрывчатым.
При действии на растворы солей серебра карбонатом натрия образуется белый осадок средней соли Ag2CO3 (200°C разл.), что свидетельствует о незначительном гидролизе ионов Ag+. Действительно, 0.1М раствор нитрата серебра имеет рН около 6. Хотя считается. Что в водных растворах ионы серебра образуют тетраэдрические аквакомплексы [Ag(H2O)4]+ (J. Texter, J.S. Hastrelter, J.L. Hall, Journ Phys. Chem., 1983, 87, 4690), ни одной соли с таким катионом до сих пор не получено. Соли серебра выделяются из водных растворов либо без молекул воды, либо в форме моногидратов (AgClO4×H2O, AgVO3×H2O). Все это свидетельствут о низком сродстве серебра к кислороддонорным лигандам – оно предпочитает более «мягкие» реакционные центры, такие как сера или азот. Так, например, оксалат серебра Ag2C2O4 в отличие от оксалатов большинства переходных металлов не растворяет при избытке оксалат-ионов, зато легко переходит в раствор в присутствии аммиака.
При действии на соли серебра аммиака или едких щелочей первые капли раствора дают беловатый осадок неустойчивого гидроксида, который сразу же превращается в бурый оксид:
2AgNO3 + 2NaOH = Ag2O¯ + 2NaNO3 + H2O.
При работе с подкисленными растворами необходимо помнить, что осадок начинает образовываться лишь в нейтральной среде, а количественно осаждается при рН 11.
Гидроксид серебра известен только в сильно разбавленных растворах, образующихся при оседании суспензии оксида Ag2O в воде:
1/2Ag2O + H2O Ag+ + OH– , K = 3,8×10–8 (25°C, 3M NaClO4)
Водная суспензия оксида серебра подобно едким щелочам поглощает из воздуха углекислый газ, превращаясь в карбонат Ag2CO3. Все это свидетельствует о том, что неустойчивый гидроксид серебра является довольно сильным основанием. Таким образом, сила гидроксидов переходны металлов последовательно возрастает по мере понижения ионного потенциала (том 2, с. 50), то есть увеличения размера (Mn2+ > Fe2+ > Co2+, Ni2+ > Cu2+ > Zn2+; Rh3+ > Co3+) и уменьшения заряда катиона (M(OH)3 < M(OH)2 < MOH).
Слабый амфотерный характер гидроксида серебра(I) проявляется в частичном растворении оксида Ag2O в крепких щелочах (pH > 13) с образованием неустойчивых дигидроксоаргентат-ионов [Ag(OH)2]–, известных лишь в растворах. Взаимодействием оксида серебра(I) с оксидами щелочных металлов получены аргентаты M4Ag4O4, M3AgO2. Они нацело гидролизуются водой.
Для серебра(I) характерны преимущественно линейные комплексы, например, дицианоаргентат(I) [Ag(CN)2]-, образующийся при растворении цианида серебра в избытке цианида калия и имеющий строение [NC–Ag–CN]-. Из водных растворов калийная соль K[Ag(CN)2] выделяется в виде бесцветных плоских шестиугольных кристаллов. Введение в раствор цианид-ионов существенно понижает окислительный потенциал ионов серебра:
[Ag(CN)2]- + e– ® Ag + 2CN–, E° = –0.29B.
Цианидные растворы, содержащие эти комплексные ионы, используют для гальванического серебрения металлических поверхностей. Это позволяет избежать реакции между ионами серебра и более активным металлом, из которого выполнено изделие. В противном случае серебро начнет выделяться в виде рыхлого осадка еще до подачи напряжения на электроды.
Линейный аммиакат [Ag(NH3)2]+ получают действием избытка раствора аммиака на соли серебра или его оксид:
Ag2SO4 + NH3 = [Ag(NH3)2]2SO4.
Соответствующее ему основание [Ag(NH3)2]OH известно лишь в растворах. В жидком аммиаке возможно образование и тетрааммикатов [Ag(NH3)4]+, подобно тому как в пиридине образуются ионы [Agpy4]+. В воде они разлагаются.
Подобно соединениям непереходных и постпереходных металлов комплексы с электронной конфигурацией d10 являются лабильными. Поэтому аммиакат легко разрушается под действием кислоты или сульфид-ионов, связывающих серебро в малорастворимый черный осадок сульфида:
[Ag(NH3)2]2SO4 + 2H2S = 2Ag2S¯ + 2(NH4)2SO4
Устойчивость галогенидных комплексов серебра(I) возрастает с увеличением порядкового номера галогена. Хлоридные и бромидные комплексы построены аналогично цианидному, то есть имеют состав [AgX2]– и линейную геометрию, в то время как в иодидных координационные числа три и четыре.
Описаны ионы [AgI3]2– , [Ag2I4]2–, [Ag3I4]–, [Ag4I5]– , [Ag4I8]4–, в зависимости от условий и природы катиона существующий в одной из трех изомерных форм, [Ag13I15]2–, [Ag18I23]5–. Интересно, что в дииодоаргентате TlAgI2 содержатся не изолированные линейные ионы, а бесконечные цепи из тетраэдров AgI4, связанных ребрами. Еще более сложные цепи из конденсированных тетраэдров обнаружены в CsAg2I3 (Рис. 7.17. Строение различных иодоаргентатов(I): (а) [AgI3]2– , (б) [Ag2I4]2–, (в) [AgI2]nn– , (г) [Ag4I8]4–, (д) [Ag2I3]nn– )
Устойчивость карбонилов металлов определяется долей p-взаимодействия, когда заполненные d-орбитали металла перекрываются с вакантными π*-орбиталями молекулы СО. Разница в энергиях взаимодействующих орбиталей возрастает в ряду Cu-Ag-Au, поэтому при пропускании угарного газа через водные растворы хлоридных комплексов этих металлов карбонилов не образуется, а происходит восстановление:
2K[AgCl2] + CO + H2O = Ag¯ + CO2 + 2HCl + 2KCl
С фосфинами галогениды серебра(I) и меди(I) образуют замкнутые кубановые или открытые ступенчатые структуры, которые в ряде случаев находятся в равновесии:
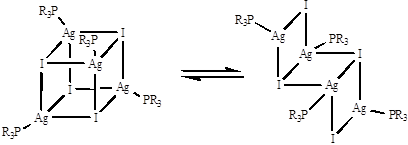
Химия золота(I) представлена бинарными галогенидами и псевдогалогенидами, а также комплексами, преимущественно аионными. В отличие от меди и серебра, оно не образует солей с оксокислотами. Причиной этого является высокое значение константы реакции диспропорционирования:
3Au+ Au3+ + 2Au, K = 1010
и большой потенциал пары Au+/Au (1.68B).
Устойчивость комплексных соединений обусловлена π-дативным взаимодействием между заполненными d-орбиталями металла и вакантными π*-орбиталями лиганда. Так как при движении вниз по группе происходит увеличение размеров d-орбиталей, степень их перекрывания с π*-орбиталями лиганда возрастает, что приводит к увеличению устойчивости комплексов (табл.7.6). В то же время в случае лигандов, не способных предоставить металлу вакантные орбитали π-типа (аммиак), устойчивость комплексов понижается по мере ослабления σ-связи металл-лиганд, то есть в ряду Cu-Ag-Au.
Таблица 7.6.
Константы устойчивости lgКуст некоторых комплексных
cоединений металлов группы меди в степени окисления +1
| Комплекс | Cu | Ag | Au |
| [MCl2]– | 5.30 | 5.40 | 9.42 |
| [MBr2]– | 5.89 | 7.11 | 12.4 |
| [MI2]– | 8.76 | 13.85 | - |
| [M(SCN)2]– | 12.11 | 7.57 | |
| [M(CN)2]– | 21.1 | 38.3 | |
| [M(NH3)2]+ | 10.86 | 7.23 | - |
| [M(S2O3)3]5- | 12.3 | 13.5 | - |
Являясь типичной мягкой кислотой, ион Au+ образует химические связи с наиболее мягкими донорными центрами лигандов: так, сульфитные, тиосульфатные и тиоцианатные группы оказываются координированы через атом серы, цианат – через атом азота.
Примером устойчивого комплексного соединения золота(I) является бесцветный дицианоаурат K[Au(CN)2], образующийся при растворении золота в растворе цианида калия в присутствии кислорода или пероксида водорода. При кипячении водного раствора соли с 2М соляной кислотой происходит разложение комлпекса, сопровождающееся выделением лимонно-желтого осадка цианида AuCN, имеющего полимерную структуру с линейными цепочками –Au-CN-Au-CN-. Методом ионного обмена из калийной соли может быть получена золотоциановодородная кислота H[Au(CN)2], которая при 100°C разлагается на цианид золота и циановодород. Растворением иодида золота(I) в растворе тиосульфата получают тиосульфатные комплексы золота(I), например, Na3[Au(S2O3)2]. Анион, входящий в его состав, имеет линейную геометрию с двумя тиосульфатными группами, координирвоанными через атом серы.
Синтез многих комплексов золота(I) проводят в неводных средах, используя азот-донорные ратсоврители, стабилизирующие металл в степени окисления +1 и тем самым смещающие равновесие диспропорционирования влево. Так, аммиакат золота [Au(NH3)2]+ получен в нитриле бензойной кислоты (D.M.P. Mingos et al, Journ. Chem. Soc., Dalton Trans., 1995, 319). Другим споосбом синтеза аммиакатов является растворение галогенидов золота в жидком аммиаке.
При растворении бромида и иодида золота в растворах соответствующих галогенидов щелочных металлов образуются линейные анионы [AuX2]–, которые критсаллизуются в виде солей с тяжелыми катионами, такими как тетраэтиламмоний. Хлорид золота(I) также растворяется в концентрированном водном растворе хлорида натрия с образованием дихлороаурата(I) [AuCl2]–, однако под действием воды этот ион легко диспропорционирует:
H2O
3[AuCl2]– ¾¾¾¾® [AuCl4]– + 2Au + 2Cl–
Галогенидные комлпексы золота(I) легко кисляются до соединений золота(III):
[AuI2]– + I2 ¾® [AuI4]– , K = 200
Карбонильный комплекс AuCOCl образуется при взаимодействии угарного газа с хлоридом золота(I) при 90°C в твердом виде или в бензольном растворе, либо при восстановлении хлорида золота(III) угарным газом:
CCl2=CCl2, 120°C
Au2Cl6 + 4CO ¾¾¾¾¾¾¾¾® 2AuCOCl + 2COCl2
Он представляет собой бесцветные кристаллы, аостроенные из линейных молекул Cl-Au-CO, сильно взаимодействующих друг с другом за счет образования дополнительныж связей Au…Cl (Рис. 7.18. Строение золотокарбонилхлорида). В отличие от аналогичного соединения меди(I) комплекс золота распадается на золото и угарный газ даже в присутствии следов влаги (Jones P., Z. fuer Naturforsch. B., 1982, 37, 823). Недавно получен и линейный дикарбонильный катион в виде соли [Au(CO)2]2Sb2F11 (H. Willner, Journ. Amer. Chem. Soc., 1992, 114, 8972). Линейная геометрия присуща и другим металлоорганическим производным золота(I): [Me2Au]– и даже [Au(acac)2]–, в котором ацетилацетонат выступает в роли углероддонорного лиганда, образующего σ-связь Au-C (J. Vicente, M.-T. Chicote, et al., Chem. Commun., 1992, 915).
Известны аураты(I) щелочных металлов (Cs4Au4O4, Na3AuO2), построенные аналогично аргентатам. Серасодержащие комплексы золота(I) входят в состав лекарственных препаратов, используемых для лечения ревматоидных артритов: преапарат ауранофин представляет собой тиоглюкозид трифенилфосфинзолота(I), а вводимый внутримышечно кризанол содержит ауротиопропанолсульфонат кальция (LY. Gade, Angew. Chem., Inern. Ed. Engl., 1997, 36, 1171).
Степень окисления +2.
В степени окисления +2 элементы группы меди имеют электронную конфигурацию d9, соответствующую ян-теллеровскому иону с искаженной октаэдрической геометрией. Соединения серебра и золота в степени окисления _2 немногочисленны и легко диспропорционируют, поэтому основное внимание в тексте уделено описанию химии меди. Ион Cu2+ в октаэдрическом поле обычно претерпевает удлинение двух связей, расположенных по оси z, приобретая геометрия квадратной бипирамиды с четырьмя лигандами, расположенными на равном расстоянии в экваториальной плоскости и двумя более отдаленными, занимающими аксиальные позиции. Координационное число, таким образом формально равно шести, но часто обозначается как 4 + 2 с целью подчеркнуть тетрагональное искажение и удаленность двух лигандов. При полном удалении этих лигандов возникает плоско-квадратная геометрия, являющаяся предельным случаем тетрагонального искажения. На практике это имеет место в случае объемных лигандов. Наличие одного неспаренного элеткрона позволяет предсказать парамагнетные свойства с магнитным моментом 1,73 М.Б., наблюдаемое на практике значение оказывается несколько выше за счет спин-орбитального взаимодействия.
Ион гексааквамеди(II) [Cu(H2O)6]2+ входит в состав многих кристаллогидратов, например, Cu(ClO4)×6H2O, (NH4)2Cu(SO4)2×6H2O, придавая им характерный для соединений меди(II) синий цвет. Из-за тетрагонального искажения электронный спектр иона в видимой области представляет собой широкую несимметричную полосу с максимумом пр 800 нм, соответствующему переходу электрона с dz2 на dx2-y2 орбиталь (Рис.7.19. Электронный спектр поглощения иона [Cu(H2O)6]2 , аммиакатов меди(II) (а) и схема расщепления d-орбиталей в тетрагонально искаженном октаэдре, удлиненном по оси z (б)). Аксиальные и экваториальные расстояния в гексаакваионе различаются примерно на 20% (Рис. 7.20. Строение иона [Cu(H2O)6]2+ в структуре перхлората меди(II) , ссылка: Galucci J.C., Gerkin R.E., Acta Cryst.C., 1989, C45, 1279). Интересно, что в структуре гексафторосиликата [Cu(H2O)6]SiF6 каждый четвертый ион [Cu(H2O)6]2+ представляет собой правильный октаэдр, но есть не испытывает ян-теллеровского искажения (F.A: Cotton et al, Inorg. Chem., 1993, 32, 4861). В структурах тетрагидратов присутствуют плоско-квадратные ионы [Cu(H2O)4]2+, дополненные до октаэдра взаимодействием с донорными атомами анионов (Рис. 7.21. Строение [Cu(H2O)4]3[FeF6]2; Сноска: Kummer S., Babel D., Z. fuer Naturforsch. B., 1987, 42, 1403)
Соли меди(II) в водных растворах гидролизованы, в них присутствуют полиядерные катионы [Cun(OH)2n-2(H2O)m]2+ с мостиковыми гидроксогруппами. Так, 0,1М ратсвор сульфата меди(II) при 15°C имеет рН 4.2. Кипячение растворов приводит к образованию малорастворимых основных солей, часто окрашенных в зеленый цвет:
t°
4CuSO4 + 6H2O ¾® Cu4(OH)6SO4¯ + 3H2SO4
Раствор при этом подкисляется.
При действии на соли меди(II) щелочей выпадает объемистый синий осадок аморфного гидроксида Cu(OH)2, который при нагревании до 125°C отщпляет воду, превращаясь в оксид CuO. Проводя синтез этого соединения, важно помнить, что даже небольшой избыток щелочи вызвает протекание дегидратации в растворе уже при комнатной температуре.
Легкость разложения гидроксида меди(II) определяется его строением, которое отлично от структуры гидроксидов других двухзарядных катионов. Упрощенно ее можно педставить как прямые цепи из квадратов Cu(OH)4 соединенные друг с другом общими сторонами. Друг относительно друга цепи расположены таким образом, что атомы кислорода из соседних цепей, находящихся сверху и снизу, дополняют координационное число атомов меди до шести (Рис. 7.22. Строение гидроксида меди(II)). Для перестройки этой структуры в оксид меди(II) требуется лишь отщепление части гидроксильных групп (оксоляция) и разворот некоторых цепей друг относительно друга.
Получение чистого кристаллического гидроксида представляет собой непростую задачу. Продукт, образующийся при действии щелочи на растворы солей меди(II) содержит примесь основных солей, а осадок, выпадающий при обратном порядке смешения реагентов быстро чернеет из-за локального избытка щелочи. По одному из методов сначала получают зеленый основной сульфат, добавляя к раствору медного купороса едкий натр в количестве 2/3 от требуемого по расчету для полного осаждения гидроксида. Затем образовавшийся яблочно-зеленый осадок обрабатывают концентрированным растовром едкого натра, взятом в стехиометрическом количестве. В результате этого образуется кристаллический гидроксид, который в отличие от аморфного имеет небесно-голубой цвет и устойчив при хранении. Его высушивают при 100°C. Ранее порошок гидроксида применяли в качестве пигмента «бременская синяя». Предложен препаративный метод синтеза, заключающийся в разложении аммиаката [Cu(NH3)4(H2O)2](OH)2 при 110°C.
Гидроксид меди(II) легко растворяется в кислотах
Cu(OH)2 + 2HCl = CuCl2 + 2H2О.
В разбавленных щелочах не растворяется, но с концентрированными растворами гидроксидов щелочных металлоа образует ярко-синие тетрагидроксокупраты (II)
Cu(OH)2 + 2NaOH = Na2[Cu(OH)4].
Это свидетельствует об амфотерности гидроксида, хотя основные свойства преобладают.
Гидроксокупраты щелочных и щелочноземельных металлов выделены в твердом виде. Фиолетовый осадок стронциевой соли Sr[Cu(OH)4]×H2O может быть получен при добавлении хлорида стронция к раствору, полученному взаимодействием бромида меди(II) с избытком 40%-ного NaOH (M.J. Pack, W. Patalinghug, M.A. Weller, Journ. Chem. Soc., Dalton Trans., 1996, 7). Ион [Cu(OH)4]2– представляет собой плоский квадрат с расстоянием Cu-O 0.193 – 0.195 нм. При разбавлении раствора гидроксокупраты гидролизуются, причем при быстром добавлении их в большой избыток воды образуется фаза гидроксида, а при медленном проведении реакции – оксид. Известны также и гексагидроксокупраты, например, Sr2[Cu(OH)6], а также оксокупраты Sr2CuO3, Ba2CuO3, Y2Cu2O5, получаемые твердофазным синтезом. Некоторые смешанные купраты(II,III) бария и редкоземельных элементов обладают высокотемпературной сверхпроводимостью.
ДОПОЛНЕНИЕ. Медный купорос
Среди солей меди(II) наибольшее значение имеет медный купорос CuSO4×5H2О, выделяющийся из водных растворов сульфата меди(II) в виде крупных сине-голубых кристаллов. В структуре этого соединения атом меди находится в плоско-квадратном окружении из четырех молекул воды. Квадраты [Cu(H2O)4]2+ соединены друг с другом η-мостиковыми сульфатными группами, расположенными сверху и снизу от плоскости каждого квадрата. Пятая молекула воды является внешнесферной - прочными водородными связями соединена не с атомом меди, а с двумя атомами кислорода сульфатных групп и двумя внутрисферных молекул воды, тем самым связывая их между собой (рис. 7.23. Строение медного купороса).
При 56°C медный купорос претерпевает полиморфный переход, при 93°C превращается в голубой тригидрат CuSO4×3H2O (Рис. 7.24. Растворимость сульфата меди(II) в воде), в структуре которого атом меди связан с координирован тремя молекулами воды, одним атомом кислорода сульфатной группы и двумя атомами кислорода двух соседних сульфатных гурпп, находящихся на более значительном расстоянии (Уэллс А. Структурная неорганическая химия, т.2, М., Мир, 1987, с. 415). Таким и образом, как и следовало ожидать, внешнесферная молекула воды, находящаяся в структуре медного купороса, при нагревании отщепляется первой. Дальнейшая дегидратация при 105°C приводит к серо-голубому моногидрату, который сохраняет устойчивость до 230°C. Выше этой температуры он полностью обезвоживается, окончательно утрачивая голубую окраску и превращаясь в светло-серый порошок безводного сульфата меди.
При растворении его в воде выделяется теплота, а раствор окрашивается в синий цвет – вновь происходит образование медного купороса:
CuSO4(тв.) + 5H2O(ж.) = CuSO4×5H2O(тв.), ΔΗ° = –1588 кДж/моль.
Это еще раз доказывает энергетическую выгодность структуры пентагидрата.
При сильном прокаливании безводный сульфат меди(II) разлагается, превращаясь в оксид:
>710°C
2CuSO4 ¾¾® 2CuO + 2SO2 + O2
В качестве побочного продукта разложения может образовываться оксосульфат Cu2OSO4, который при 860°C превращается в оксид меди(II), сернистый газ и кислород. Препаративным методом получения безводного сульфата также является взаимодействие хлорида меди(II) с серным ангидридом в жидком сернистом газе или хлористом сульфуриле:
CuCl2 + 2SO3 = CuSO4 + SO2Cl2.
Медный купорос используется в электрохимических процессах нанесения медных покрытий, для консервации древесины, протравливания семян, в производстве минеральных красок и как исходное вещество для синтеза различных соединений меди. Суспензию гидроксида кальция в растворе сульфата меди(II) (бордосская жидкость) применяют для защиты растенийот вредителей и болезней. В технике медный купорос получают растворением медных отходов в горячей концентрированной серной кислоте или обработкой подвергнутых обжигу сульфидных руд теплой разбавленной серной кислотой.
Кипячение растворов сульфата меди(II), как было указано выше, приводит к выпадению зеленого осадка основного сульфата Cu4(OH)6SO4, встречающегося в природе в виде минерала брошантита. Эту соль проще приготовить взаимодействием водной суспензии гидроксида с раствором сульфата меди(II). Известен также его гидрат Cu4(OH)6SO4×H2O – минерал познякит, окрашенный в голубой цвет. Он кристаллизуется со временем из раствора медного купороса, в который был добавлен гидрокарбонат натрия до рН 6.5 (M.M. Naumova, S.A: Pisareva, G.O. Nechiporenko, Studies in conservation, 1990, 35, 81).С сульфатами щелочных металлов медный купорос дает двойные соли – шениты («соли Туттона»), например, K2SO4×CuSO4×6H2O. Они имеют более низкую растворимость по сравнению с исходными веществами и легко кристаллизуются.
КОНЕЦ ДОПОЛНЕНИЯ
Нитрат меди(II) ниже 24,5°C кристаллизуется из водных растворов в виде синего гексагидрата Cu(NO3)2×6H2O, содержащего ионы гексааквамеди(II); выше этой температуры устойчив гидрат Cu(NO3)2×3H2O. Осторожной дегидратацией в вакууме 0,0001 атм при 60 – 70°C в течение нескольких его удается перевести в моногидрат, а повышая температуру до 80 – 90°C и окончательно обезводить. Тем не менее, наиболее удобен синтез безводного нитрата из меди действием на нее жидким N2O4 растворенным в этилацетате (том 2, с. 192):
Cu + 2N2O4 = [NO]+[Cu(NO3)3]- + 2NO.
Образующуюся соль нитрозония высаливают избытком диоксида азота, а затем разлагают в вакууме
100°C
[NO]+[Cu(NO3)3]- ¾¾® Сu(NO3)2 + 2NO2.
Синие кристаллы безводного нитрата меди(II) плавятся при 147оС, а при 160°C возгоняются, переходя в пар в форме мономерных молекул Сu(NO3)2 (рис. 7.25. Строение нитрата меди(II): (а) в парах, (б) в кристалле). В твердой фазе вещество существует в виде двух полимофрных форм (Троянов С.И., Морозов И.В., Знаменков К.О., Коренев Ю.М. Ж. неорганической химии, 1996, т. 41, № 9, стр. 1476-1483), построенных из атомов меди, объединенных мостиковыми нитратными группами объединяются в каркас из бесконечных зигзагообразных цепей (рис. б).
При смешении раствора нитрата меди(II) с суспензией гидроксида Cu(OH)2 образуется основная соль Cu2(NO3)(OH)3, найденная в в природе в виде минерала герхардтита.
Средний нитрит меди(II) не известен. При попытке концентрирования раствора, содержащего образующие его ионы, происходит диспропорционирование нитрита на нитрат и NO. Взаимодействием ПОСМОТРЕТЬ СТАТЬЮ может быть получена основная соль Cu2(OH)3(NO2) в виде игольчатых кристаллов, растворимых в воде и спирте (Schmidt M., Moeller H., Lutz H.D., Z. Anorg. Allg. Chem., 1993, 619, 1287). Извсетны также многочисленные нитритные комплексы, в которых нитрит-ион координирован через атом азота: K3[Cu(NO2)5], [Cu(NH3)4(NO2)2], [Cu(NH3)2(NO2)2] (Rochdi K,m Riou A., Cudennec Y., et al, Eur. Journ. Solid State Chem., 1993, 30, 427 и 1143).
Описано большое количество фосфатов меди(II). Синий ортофосфат Cu3(PO4)2 взаимодействует с форсфорной кислотой с образованием кислых солей CuHPO4 и Cu(H2PO4)2 (Boudjada A., Mater. Res. Bull., 1980, 15, 1083). Получены основные соли Cu3(PO4)(OH)3, Cu2(PO4)(OH) и Cu5(PO4)2(OH)4, использовавшийся в качестве природного зеленого пигмента («псевдомалахит»), оксофосфаты Cu4(PO4)2O, Cu5(PO4)2O2, пиро- Cu2P2O7 и тетрамета- Cu2P4O12 фосфат. Известны средний CuHPO3(H2O)2 и кислый Cu(H2PO3)2 фосфиты меди(II).
При смешении растворов арсенита натрия и сульфата меди(II) выпадает зеленый осадок переменного состава, ивестный как зелень Шееле. Он представляет собой основный арсенит меди(II), точный состав и строение которого однозначно не установлены. Растворением зелени Шееле в кипящей уксусной кислоте получали другой зеленый пигмент – швейнфуртскую зелень Cu(CH3COO)2×3Cu(AsO2)2. В настоящее время эти вещества не используют из-за их высокой токсичности.
При взаимодействии солей меди с жидким стеклом выпадают осадки силикатов. Известны структуры солей цепочечного метасиликата CuSiO3(H2O), встречающегося в природе в виде минерала диоптаза, а также основных солей Cu5(OH)2(SiO3)4, Cu5(Si4O12)(OH)2.
Оксалаты щелочных металлов осаждают из растворов солей меди(II) голубой осадок оксалата CuC2O4×H2O, растворимый в избытке реагента с образованием комплексов [Cu(C2O4)2]2–, а при нгревании разлагающийся на воду, медь и углекислый газ.
Слабые основные свойства гидроксида меди(II) наглядно демонстрирует невозможность синтеза в водном растворе некоторых средних солей меди(II) со слабыми кислотами, например, карбоната. В результате взаимодействия растворов сульфата меди(II) и гидрокарбоната натрия в зависимости от соотношения реагентов образуется голубой осадок основного сульфата (при большом избытке сульфата меди) либо (при мольном соотношении реагентов 1 : 3) светло-зеленый осадок гидроксокарбоната меди(II) Cu2(OH)2CO3 – малахита, или горной зелени:
2CuSO4 + 4NaHCO3 = Cu2(OH)2CO3¯ + 2Na2SO4 + 3CO2 + H2O
Строго говоря, образующийся осадок представляет собой гидрат Cu2(OH)2CO3×0.5Н2О со структурой малахита. При нагревании он обезвоживается. В природе также встречается основная соль Cu3(OH)2(CO3)2 синего цвета – минерал азурит (“медная лазурь”, горная синь). Удобного метода синтеза этого вещества пока не разработано.
Средний карбонат меди(II) получают взаимодействием оксида, гидроксида или основного карбоната меди с углекислым газом при высоком давлении:
20 кбар, 500оС
Cu2(OH)2СО3 + СО2 ¾¾¾¾¾¾® 2CuСО3 + Н2О.
Структура CuСО3 построена из квадратно-пирамидальных полиэдров [CuO5], в которых атомы находятся в окружении атомов кислорода пяти различных карбонатных групп (Seidel H., Viswanathan K., Johannes W., Ehrhardt H., Z. Anorg. Allg. Chem., 1974, 410, 138). При кипячении суспензии малахита в концентрированном растворе соды происходит образование синего раствора, из которого кристаллизуется голубой карбонатокупрат Na2[Cu(CO3)2] (Пупкин, Inorg. Chem., 1981, 20, 78)
Осадок малахита легко растворим в кислотах с выделением углекислого газа,в растворе аммиака с образованием синих аммиакатов, а при нагревании рзлагается:
250°C
Cu2(OH)2CO3 ¾¾¾¾¾® 2CuO + CO2 + H2O
Окислительные свойства ионов меди(II) могут быть обнаружены при внесении в раствор соли сильного восстановителя: металла, более активного чем медь или альдегида:
Fe + CuSO4 = FeSO4 + Cu
Наиболее ярко окислительные свойства выражены в щелочной среде, однако студенистый осадок гидроксида меди(II) неудобно использовать в качестве реагента. Замечено, что в присутствии винной, лимонной или яблочной кислот едкие щелочи не осаждают из раствор медного купороса гидроксид меди(II), так как ионы меди связаны в устойчивые комплексы. В то же время такой раствор проявляет окислительные свойства, чем пользуются в лабораторной практике для обнаружения альдегидной группы, гидразина, гидроксиламина и других восстановителей. Обычно применяют жидкость Фелинга – щелочной раствор медного купороса, содержащий винную кислоту. Ее готовят смешивая равные объемы раствора, содержащего 34,65 г сульфата меди(II) в 500 мл воды и раствора, полученного растворением 173 г сегнетовой соли (тартрата натрия-калия) и 52 г гидроксида натрия в 500 мл воды. Оба раствора хранят отдельно и смешивают непосредстивенно перед употреблением. О наличии восстановителя свидетельствует образование желтого или оранжевого осадка оксида меди(I) (см. выше).
Медь(II), подобно другим металлам группы, тяготеет к мягким донорным центрам – азоту и сере. При движении по 3d-ряду легкость образования аммиакатов и их учтойчивость полседовательно возрастают. Так, лазурно-синие аммиачные комплексы меди(II) образуются при действии на соли меди(II) избытком водного раствора аммиака:
Cu2+ + 4NH3 + 2H2O = [Cu(NH3)4(H2O)2]2+
Процессы замещения молекул воды в координационной сфере меди на молекулы аммиака протекают последовательно, но с высокой скоростью, обусловленной лабильностью комплексов с d9-конфигурацией атома металла. По мере протекания замещения происходит увеличение параметра расщепления Δo, что приводит к последовательному смещению полосы поглощения в коротковолновую часть спектра и изменению окраски от сине-голубой до фиолетовой (см. рис. 7.19) . При избытке аммиака в водных растворах преобладают тетраммианокомлпексы, в которых четыре млекулы аммиака находятся в одной плоскости, а молекулы воды, расположенные сверху и снизу, дополняют геометрию до тетрагонально искаженного октаэдра. Кристаллизация соли, которую легко достичь добавлением в раствор спирта или ацетона, приводит к отщеплению одной молекулы воды и образованию мостиков [Cu(NH3)4(μ2-H2O)](SO4) (Рис. 7.26. Строение тетрааммиакатов меди(II) в кристаллических солях: [Cu(NH3)4(μ2-H2O)](SO4) (а) и [Cu(NH3)4(MnO4)2] (б)). В других случаях происходит замещение молекул воды на анион. Ионы [Cu(NH3)5(H2O)]2+ начинают преобладать лишь при концентрации аммиака около 10 моль/л, а гексамминокомлпексы образуются лишь в отсутствие воды (Рис. 7.27. Зависимость содержания различных аммино-комплексов меди(II) в водных растворах от молярной концентрации аммиака). Трудность образования этих ионов, наглядно выраженная в последовательном понижении констант устойчивости в ряду K([Cu(NH3)4(H2O)2]2+) > K([Cu(NH3)5(H2O)]2+ ) > K([Cu(NH3)6]2+) объясняется удаленностью аксиальных положений вследствие эффекта Яна-Теллера.
При действии газообразного аммиака на безводный хлорид меди(II) ии его раствор в этилацетате может быть получен гексааммин [Cu(NH3)6]Cl2, который при растворении в воде превращается в тетрааммин. Если вместо хлорида меди(II) взять безводный сульфат, синтез приводит к [Cu(NH3)5SO4], который при нагревании теряет одну молекулу аммиака. Ион пентаамминмеди(II) в K[Cu(NH3)5](PF6)3 имеет квадратно-пирамидальную геометрию (Пупкин, Journ. Chem. Soc., Dalton Trans., 1983, 645).
Аммиакаты Cu(II) постепенно разрушается при кипячении с раствором щелочи и быстро – при пропускании сероводорода из-за образования малорастворимого сульфида:
[Cu(NH3)4(H2O)2]SO4 + 3H2S = CuS¯ + (NH4)2SO4 + 2NH4HS + 2H2O,
Многие комлпексы с серасодержащими лигандами при нагревании претерпевают внутримолекулярное окисление-восстановление. Например, при действии тиосульфата на раствор сульфата меди(II) наблюдается обесцвечивание, вызванное образованием комплексов [Cu(S2O3)(H2O)4] и [Cu(S2O3)2(H2O)2]2–. Кипячение раствора приводит к разрушению комплекса и выпадению красновато-бурового осадка:
2Cu2+ + 2S2O32– + 2H2O = Cu2S¯ + 2HSO4– + 2H+ + S¯
Среди комплексов с кислороддонорными лигандами наиболее устойчивы хелаты, например, темно-синий ацетилацетонат Cu(acac)2, образующийся при действии ацетилацетона на ацетат меди(II). В твердом виде вещество построено из плоских молекул (Рис. 7.28. Строение ацетилацетоната меди(II): (а) молекула, (б) упаковка).Благодаря молекулярному строению ацетилацетонат может быть очищен сублимацией. Интересно, что при использовании дикетонов с акцепторными группами, ослабляющими связь Cu-O, из растворов образуются зеленые гидраты состава, например, Cu(hfa)2(H2O), где Hhfa – 1,1,1,5,5,5-гексафторацетилацетон. При кипячении дикетонатов меди в спиртовом растовре щелочи происходит замещение одного лигандов на алкоксидные группы, связвающие атомы меди в димеры (acac)Cu(μ2-OR)2Cu(acac). Алкоголяты меди(II) Cu(OR)2 получают действием на хлорид меди(II) избытком алкоголята лития.
Получение фталоцианина меди(II) нагреванием соли меди(II) с динитрилом фталевой (1,2-бензолдикарбоновой) кислоты или смесью фталевого ангидрида с мочевиной при 250 – 300°C является примером темплатного синтеза (см. главу 1). Образующийся пигмент («монастраль синий») представляет собой внутрикомплексное соединение, в котором атом меди находится в плоско-квадртаном окружении из четырех атомов азота. Он выдерживает нагревание до 500°C, устойчив к действию света и щелочей. Фталоцианин меди применяют для производства типографских и художественных красок, эмалей, нитроэмалей, для окрашивания каучука, линолеума, бумаги. Применение находят также хлорированный и сульфированный фталоцианины меди(II), окрашенные в зеленые тона различных оттенков.
ДОПОЛНЕНИЕ. Карбоксилаты меди(II).
Моногидрат ацетата меди(II) Cu(CH3COO)2×H2O кристаллизуется из растворов, полученных взаимодействием основного карбоната или гидроксида со стехиометрическим количеством уксусной кислоты или по реакции ионного обмена между растворами сульфата меди(II) и ацетета бария. Вещество представляет собой темные сине-зеленые призматические кристаллы, хорошо растворимые не только в воде, но и спирте, эфире. Все это указывает на молекулярное строение вещества. В твердом виде и в растворах в органических растворителях оно состоит из диядерных кластеров Cu2(CH3COO)4(H2O)2 с четырьмя η2-ацетатными группами, образующими симметричную структуру, отдаленно напоминющую «китайский фонарик» (Рис. 7.29. Ацетат меди(II): (а) строение кластера Cu2(CH3COO)4(H2O)2, (б) образование σ- и δ-связей Cu-Cu при перекрывании d-орбиталей; (в) антиферромагнитное взаимодействие неспаренных электронов в диядерном кластере). Молекулы воды занимают боковые положения и при перекристаллизации из донорных растворителей могут быть замещены на спирт, пиридин, хинолин, пиразин, диметилфорамид (V.M. Rao, D.N. Sathyanarayana, H. Manohar, Journ. Chem. Soc., Dalton Trans., 1983, 2167). Расстояние Cu-Cu в ацетатате (0.262 нм) лишь ненамного превосходит аналогичное расстояние в металлической меди, что свидетельствует о частичном перекрывании dz2 орбиталей (σ-связь) и dx2-y2 орбиталей (δ-связь). Помимо этого, предполагается обменный механизм взаимодействия посредством π-орбиталей мостиковых ацетатных групп. Магнитный момент вещества оказывается пониженным по сравнению с рассчитанным исходя из наличия у каждого атома меди одного неспаренного электрона, что свидетельствует об антиферромагнитном взаимодействии двух неспаренных электронов. При этом происходит расщепление энергии на два уровня – нижний снглетный с нулевым суммарным спином, который соответствует димагнитному сотоянию и верхний триплетный со спином равным единице и характеризующийся парамагнетизмом. При 0°C энергетическая разница между обоими состояниями оценивается в 2,5 кДж/моль, а при понижении температуры она резко возрастает, приводя к увеличению заселенности синглетного состояния, уменьшению магнитной восприимчивости и появляению димагнетизма.
При нагревании в вакууме вещество дегидратируется, а затем переходит в газовую фазу.
При длительном взаимодействии меди с раствором уксусной кислоты в присутствии кислорода воздуха образуется зеленовато-синий порошок основного ацетата Cu2(OH)2(CH3COO)2×5H2O, используемый в качестве зеленого пигмента под названием медянка. Ранее для получения этой соли медные полосы пересыпали винограднымивыжимками, при брожении которых образуется уксусная кислота. По истечении нескольких недель образующийся налет соскабливали. В настоящее время медянку получают нейтрализацией гидроксида меди(II) стехиометрическим количеством уксусной кислоты.
КОНЕЦ ДОПОЛНЕНИЯ
Крупные анионные лиганды, такие как хлорид и особенно бромид способны образовывать с медью(II) тетраэдрические комплексы [CuX4]2–. Однако такая геометрия не позволяет достичь большого энергетического выигрыша, поэтому в кристаллах отдельные тетраэдрические анионы стабилизируются лишь в присутствии крупных катионов, таких как ион цезия или тетраметиламмония, которые препятствуют их взаимодействию друг с другом. Уже при замене цезия рубидием структура тетрахлорокупрата меняется – вместо изолированных тетраэдрических ионов внем присутствуют искаженные октаэдры CuCl6, соединенные общими вершинами (Рис. 7.30. Строение хлорокупратов(II): (а) Rb2CuCl4, (б) KCuCl3 – отдельный анион и взаимодействие между анионами в кристалле, в обоих случаях катионы не показаны). Аммонийная соль (NH4)2CuCl4 содержит изолированные анионы, которые имеют не тетраэдрическую, а плоско-квадратную геометрию. Изменение геометрии иона сказывается и на окраске соединений: плоско-квадратные комплексы имеют зеленую окраску, а тетраэдрические – оранжевую.
При кристаллизации из водных растворов могут быть получены зеленые дигидраты, например, K2[CuCl4(H2O)2], в которых плоскоквадратная геометрия аниона дополнена до искаженной октаэдрической двумя молекулами воды, расположенными в аксиальных положениях. Таким образом, в водном растворе существует равновесие
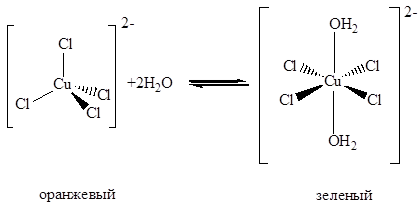
При добавлении хлорида щелочного металла в водный раствор хлорида меди(II) могут быть выделены также красные комплексы состава MCuCl3, содержащие плоские анионы, состоящие из двух квадратов CuCl4, соединенных общей стороной:
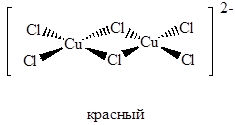
Эти ионы входят в состав большинства трихлорокупратов. Исключение составляет цезиевая соль CsCuCl3, в структуре которой присутствуют окта
Дата добавления: 2016-01-03; просмотров: 5615;