Острое реперфузионное повреждение миокарда
Ранняя реперфузия ишемизированного миокарда представляет собой общепринятый подход к терапии острых коронарных синдромов. Реперфузия миокарда после кратковременной ишемии (длящейся до 15 минут) приводит в эксперименте к полному восстановлению сократимости без каких-либо морфологических проявлений повреждения ткани. Реперфузия, наступающая после более длительной ишемии, сопровождается развитием феномена, называемого реперфузионным повреждением миокарда.
Исторически представление о природе реперфузионного повреждения миокарда развивалось на основе выдвинутой в конце 70-х годов (Hearse и соавт., 1978) концепции “кислородного парадокса”. Авторы этой концепции наблюдали ультраструктурные изменения миокарда и повышение биохимических маркеров цитолиза при реоксигенации изолированного сердца крысы после периода аноксии. Реперфузионное повреждение миокарда разделяют на обратимое и необратимое (летальное), что определяется продолжительностью предшествующего ишемического эпизода. В случае, если реперфузия наступила в течение 20 минут с момента начала ишемии, как правило, наступает обратимое угнетение сократимости миокарда, называемое персистирующей постишемической дисфункцией или станнированием. При реперфузии миокарда после 30 минут ишемии, наступает летальное реперфузионное повреждение, проявляющееся расширением зоны некроза миокарда. В настоящее время под летальным реперфузионным повреждением миокарда понимают гибель кардиомиоцитов, сохранивших жизнеспособность к моменту начала реперфузии, под действием факторов, вызванных реперфузией. Летальное реперфузионное повреждение миокарда подразделяют на острое или немедленное, приводящее к гибели кардиомиоцитов в течение первых минут реперфузии, и отсроченное летальное повреждение, возникающее вследствие активации комплемента и полиморфноядерных лейкоцитов, а также индукции апоптоза кардиомиоцитов. Еще один значимый аспект реперфузионного повреждения миокарда - возникновение индуцированных реперфузией тахиаритмий.
В противоположность медленной, прогрессивной гибели кардиомиоцитов в ходе ишемии, острое реперфузионное повреждение носит “взрывной” характер и развивается в течение нескольких минут после начала реперфузии. Выделяют четыре важнейшие причины наступления острого реперфузионного повреждения:
1) массивное образование повреждающих концентраций свободных радикалов кислорода и продуктов перекисного окисления липидов,
2) развитие гиперконтрактуры кардиомиоцитов вследствие быстрого восстановления энергетического потенциала клетки,
3) усиление гиперконтрактуры из-за быстрой нормализации тканевого значения рН,
4) отек кардиомиоцитов в результате осмотической перегрузки.
Роль свободных радикалов. Реперфузионное повреждение ткани под действием свободных радикалов кислорода возникает в том случае, когда выраженность оксидативного стресса превосходит кислородсвязывающую емкость эндогенных антиоксидантных систем. Восстановление молекулярного кислорода приводит к образованию супероксиданион-радикала (О2-). Образование супероксиданион-радикала представляет собой первый этап формирования другихактивных форм кислорода, таких как перекись водорода и гидроксил-радикал (·ОН). В реакции Хабер-Вейсса в ходе взаимодействия О2- с перекисью водорода образуется молекулярный кислород и два гидроксил-радикала. В реакции Фентона гидроксил-радикал образуется при взаимодействии двухвалентного железа с перекисью водорода. В последнее время большое внимание уделяют образованию другого свободного радикала - пероксинитрита (ONOO-), вносящего существенный вклад в реперфузионное повреждение.
Пероксинитрит-радикал образуется при взаимодействии супероксиданион-радикала и оксида азота (NO). При протонировании ONOO- образуется пероксиазотная кислота (ONOOH). Пероксиазотная кислота при ее спонтанном разложении может являться дополнительным источником гидроксил-радикалов и диоксида азота (NO2).
Активные формы кислорода, взаимодействуя с фосфолипидами сарколеммы кардиомиоцитов и образуя короткоживущие соединения – липоперекиси, приводят к повышению жидкостности мембраны, дестабилизируют ее и способствуют ее разрыву, действуя совокупно с другими повреждающими факторами реперфузии.
Быстрое восстановление энергетического потенциала клетки (рис. 8). Восстановление доставки кислорода и основных субстратов энергетического обмена кардиомиоцитов после периода ишемии парадоксально приводит к летальному повреждению клеток, способствуя возникновению гиперконтрактур, что морфологически проявляется наличием “поясков сокращения” (contraction bands) в зоне некроза. Механизм формирования гиперконтрактуры при реперфузии включает восстановление энергетического обеспечения катионных насосов и активацию сократительного аппарата кардиомиоцитов за счет доставки АТФ к его миофибриллярным элементам.
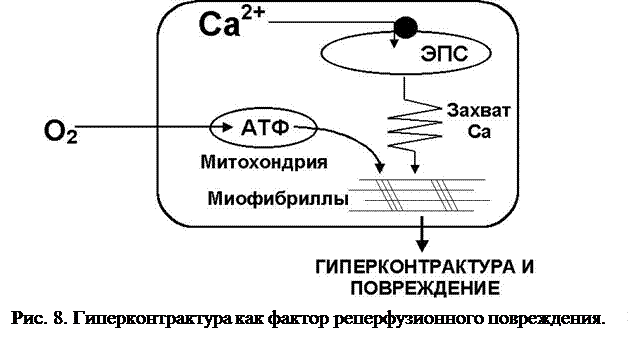 |
В состоянии острого энергодефицита, вызванного ишемией, наблюдается повышение концентрации ионов натрия (Na+) и кальция (Ca2+) в саркоплазме кардиомиоцитов. Таким образом, в начальной стадии реперфузии внутриклеточная концентрация Ca2+ остается достаточно высокой, что при восстановлении энергетического потенциала создает предпосылку для неконтролируемого, избыточного сокращения клеток. Постоянное сокращение вызывает гиперконтрактуру. В ткани миокарда, представляющей собой функциональный синцитий, гиперконтрактуры кардиомиоцитов неизбежно приводят к разрыву мембран соседних клеток и их гибели.
Значение быстрой нормализации рН (рис. 9). Значение рН в цитоплазме кардиомиоцита после реперфузии оказывает существенное влияние на развитие гиперконтрактуры. После продолжительной ишемии внутриклеточное значение рН резко снижено вследствие анаэробного метаболизма, сопровождающегося накоплением лактата. Это приводит к закислению как внутриклеточного сектора ткани, так и ее интерстиция. При реперфузии в интерстиции быстро нормализуется значение рН, в то время как внутриклеточная рН еще остается сниженной и возникает транссарколеммальный градиент рН. Это приводит к активации механизмов выведения Н+ из клетки, а именно, Na+/H+ обменника и Na+/HCO3- симпортера. Этот процесс имеет два важных последствия: снижение внутриклеточного ацидоза и дополнительный приток Na+ в цитоплазму. При вызванной ишемией несостоятельности сарколеммальной Na+,K+-АТФазы и невозможности выведения избытка Na+ из цитоплазмы, это может приводить к вторичной активации Na+/Ca2+ обменного механизма, транспортирующего Na+ из клетки, а Ca2+ - внутрь клетки (“обратный” вариант работы насоса), что усиливает предсуществующую перегрузку кардиомиоцитов кальцием.
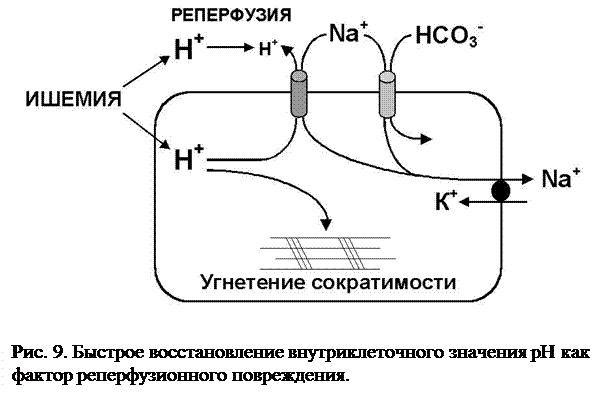 |
Таким образом, быстрое удаление протонов равно как и вторичная кальциевая перегрузка способствуют развитию гиперконтрактуры в случае, если клетка нормализует свое кислотно-основное состояние в короткий срок.
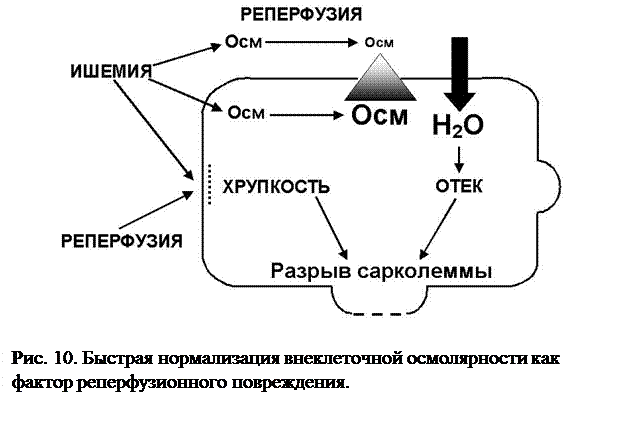
Значение быстрой нормализации осмолярности (Рис. 10). Одной из важнейших причин поступления воды в кардиомиоцит, подвергшийся ишемии/реперфузии, является перегрузка цитозоля натрием. Na+/H+ обменник играет исключительно важную роль в регуляции объема клетки. В ишемизированном миокарде также накапливаются конечные продукты анаэробного метаболизма, что дополнительно способствует гиперосмолярности вне- и внутриклеточного секторов. При реперфузии наступает быстрое удаление осмотически активных веществ из интерстиция и поэтому между вне- и внутриклеточным пространством формируется значительный осмотический градиент. Это приводит к поступлению в клетку воды, повышению внутриклеточного давления и механическому растяжению сарколеммы. Отек, в отличие от гиперконтрактуры, не способен привести к разрыву сарколеммы, что подтверждается сохранением целостности сарколеммы изолированных кардиомиоцитов, подвергнутых осмотическому стрессу при нормоксии. Однако, действуя совместно с другими факторами реперфузионного повреждения, осмотическая перегрузка клетки может способствовать ее гибели.
 |
Механизм вызванной ишемией хрупкости сарколеммы остается до конца не выясненным. В происхождении этого феномена может играть роль изменение липидного состава сарколеммы, модификация сарколеммальных протеинов, а также изменение связей сарколеммы и цитоскелета. Кроме того, имеются данные об усилении восприимчивости сарколеммы к механическим воздействиям в ходе первых минут реперфузии. Охарактеризованные выше четыре важнейших механизма реперфузионного повреждения миокарда тесно взаимосвязаны между собой. Так, перегрузка клетки кальцием, возникающая по времени раньше, способна усиливать образование свободных радикалов во время реперфузии. С другой стороны, образование активных форм кислорода может предшествовать кальциевой перегрузке и облегчать поступление Ca2+ в клетку. Например, активные формы кислорода вызывают дисфункцию саркоплазматического ретикулума, затрудняя секвестрацию Ca2+ из сарколеммы. Кроме того, сарколеммальная Na+,К+-АТФаза исключительно чувствительна к повреждающему воздействию свободных радикалов, а инактивация этого фермента является важнейшей причиной усугубления кальциевой перегрузки кардиомиоцита во время реперфузии.
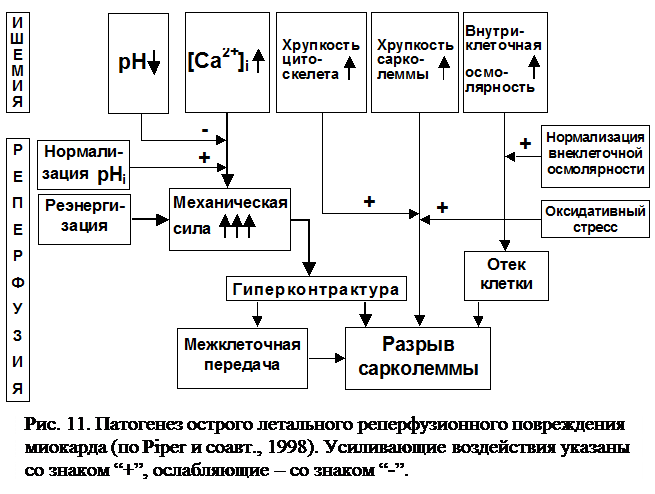
Таким образом, механический разрыв сарколеммы является непосредственной причиной острого летального реперфузионного повреждения, причем ведущее значение при этом имеет формирование гиперконтрактуры миофибрилл. Гиперконтрактура становится возможной при “реэнергизации” ишемизированной клетки, когда на фоне кальциевой перегрузки и возросшей ломкости элементов цитоскелета генерируется избыточная механическая сила. Ишемический ацидоз угнетает активацию сократительного аппарата. Резкое восстановление тканевого рН действует как разрешающий фактор для гиперконтрактуры и, помимо этого, способствует дальнейшей перегрузке кальцием. Отек клетки и повышение внутриклеточного давления, вызванные быстрой нормализацией внеклеточной осмолярности, дополнительно способствуют разрывам сарколеммы (рис. 11).
Таким образом, реперфузионное повреждение миокарда является самостоятельным патогенетическим фактором, усугубляющим ишемическое повреждение.
Дата добавления: 2015-09-18; просмотров: 2534;