Основные представления о патогенезе ишемического повреждения сердца
Ишемия миокарда возникает вследствие нарушения соответствия между величиной коронарной перфузии и метаболическими потребностями миокарда. Резкое снижение кровотока в сердце приводит к прогрессирующим метаболическим расстройствам в кардиомиоцитах. Последовательность внутриклеточных событий, развивающихся при острой ишемии миокарда, традиционно рассматривают в виде временной шкалы с соответствующими определенному времени процессами (“ишемический каскад”, схема 2).
Активация гликолиза и гликогенолиза. В течение нескольких секунд с момента наступления полной ишемии прекращается транспорт электронов по дыхательной цепи митохондрий. Одновременно активируются гликогенолиз и анаэробный гликолиз посредством активации ключевых ферментов этих процессов, - фосфорилазы и фосфофруктокиназы, соответственно. Гликогенолиз усиливается под действием освобождающихся из ишемизированных нервных окончаний катехоламинов. В результате активации гликолиза возрастает внутриклеточная концентрация НАДН, лактата и Н+, что вызывает ингибирование глицеральдегид-3-фосфатдегидрогеназы. Таким образом, по мере нарастания тяжести ишемии активность гликолиза снижается. Запасы высокоэнергетических фосфатов в миокарде ограничены и составляют ~ 20 μМ/г. Этого количе 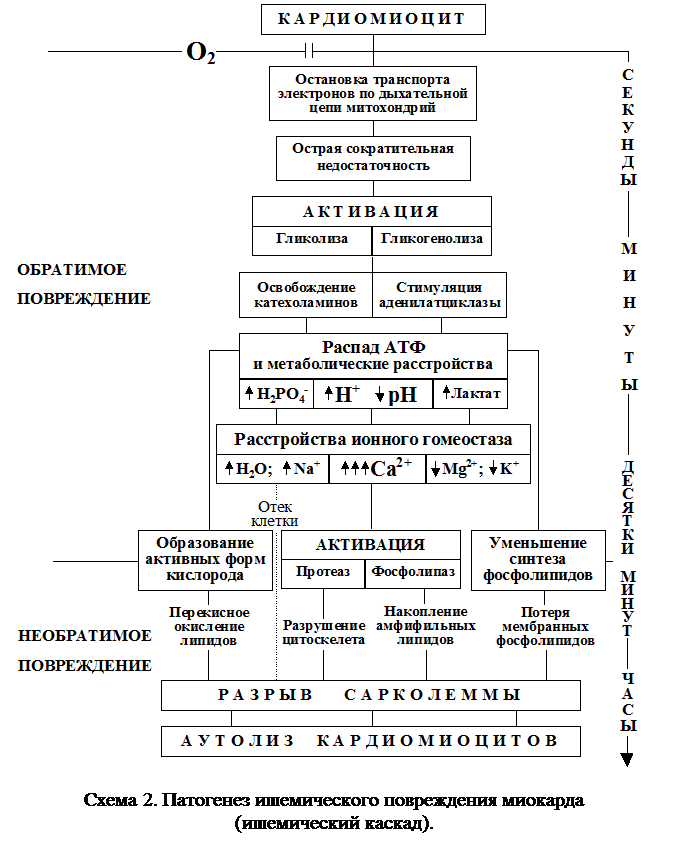
ства достаточно для поддержания нормальной сократимости миокарда в течение менее чем 30 секунд (при полной остановке притока крови).
При острой ишемии потребность в высокоэнергетических фосфатах многократно превышает скорость их образования, единственным источником которого является гликолиз, что быстро приводит к снижению содержания АТФ в миокарде и нарушению его сократимости. При этом накапливаются конечные продукты метаболических процессов: лактат, протоны водорода, неорганический фосфат и пуриновые основания, образующиеся в результате распада адениннуклеотидов. Совместно эти метаболиты создают значительную осмотическую нагрузку в клетке. Развитие внутриклеточного ацидоза вызывает вторичное ухудшение сократительной функции миокарда вследствие вытеснения Са2+ из актомиозиновых комплексов, угнетения ключевых ферментов гликолиза и ультраструктурных изменений, включающих уплотнение ядерного хроматина и образование аморфных уплотнений в митохондриальном матриксе.
Нарушения ионного гомеостаза кардиомиоцитов. Одно из проявлений этих нарушений - потеря внутриклеточного калия с одновременным возрастанием его внеклеточного содержания. Возможными причинами являются угнетение Na+/K+-АТФазы, активация АТФ-чувствительных К+ каналов, а также повышение осмотического давления внутри клетки, что приводит к выходу К+ и соответствующих анионов (лактата и фосфата).
Кроме изменения содержания К+, в раннем периоде ишемии в кардиомиоцитах происходит накопление Са2+. Механизмы увеличения концентрации Са2+ включают его поступление в клетку по потенциал-зависимым Са2+-каналам, а также возрастание его притока в клетку по обменным сарколеммальным насосам. Внутриклеточный ацидоз приводит к активации Na+/H+-обменника, выводящего из клетки Н+ в обмен на Na+. В свою очередь, это приводит к активации Na+/Ca2+ обмена. Последствиями возрастания внутриклеточной концентрации Са2+ являются активация протеаз, липаз и фосфолипаз, ингибирование биологического окисления в митохондриях и их повреждение, а также ишемическая контрактура кардиомиоцитов. В клетке происходит также накопление неорганического фосфата, образующегося в результате распада АТФ.
Электрофизиологические нарушения при ишемии включают укорочение потенциала действия, замедление проводимости, дисперсию рефрактерности и соответствующие изменения электрокардиограммы. Комплекс ионных нарушений представляет собой клеточную основу возникновения нарушений ритма – событий, часто сопровождающих раннюю фазу ишемического повреждения.
Метаболические нарушения в ишемизированном миокарде. При выраженной ишемии в миокарде угнетается митохондриальное окисление жирных кислот (ЖК) и основным субстратом энергетического метаболизма становится глюкоза. Однако при ишемии средней тяжести, например, при сохраняющемся коллатеральном кровотоке, происходит усиление захвата ЖК в зоне ишемии. Поступление ЖК при этом превышает скорость их утилизации, что приводит к накоплению избытка свободных ЖК и их эфиров. Эти соединения в высоких концентрациях обладают выраженным повреждающим действием в отношении сарколеммы и митохондрий. В ишемизированном миокарде наблюдается активация клеточных протеаз, имеющая целый ряд отрицательных последствий, включающих, в частности, деградацию белков цитоскелета.
Нарушение целостности клеточных мембран. Критическим событием в патогенезе ишемического повреждения сердца является переход обратимой стадии повреждения в необратимую. При критической ишемии (снижении кровотока в одной из магистральных коронарных артерий более чем на 80%) необратимое повреждение у человека начинается в среднем с 20 минуты и приобретает выраженный характер с 30 минуты. Необратимость повреждения связана с метаболическими расстройствами, индуцированными на ранних стадиях ишемии. Наиболее важным механизмом, ответственным за прогрессирование повреждения, в настоящее время признается нарастающее нарушение целостности клеточных мембран. По меньшей мере четыре важнейших пути ведут к потере целостности мембран:
· деградация мембранных фосфолипидов;
· образование амфифильных липидов и их интеграция в липидный бислой с изменением его жидкостности;
· образование активных форм кислорода (АФК), ведущее к перекисному окислению мембранных липидов;
· повреждение цитоскелета (включая места прикрепления элементов цитоскелета к мембранным фосфолипидам) кальций-активируемыми протеазами.
Одновременно происходит нарастание отека клетки из-за поступления в нее Na+ и воды. Продолжающееся накопление Na+ и воды на фоне серьезных метаболических расстройств и нарушения взаимосвязи цитоскелета и поверхностного аппарата клетки в конце концов приводит к разрыву сарколеммы как конечному этапу прогрессии необратимого повреждения.
Распространение некроза в ишемизированном миокарде происходит в виде “волны клеточной гибели”, зарождающейся в субэндокарде и постепенно, с течением времени распространяющейся по направлению к эпикардиальной поверхности. Данный феномен объясняется тем, что внутренние слои миокарда наиболее удалены от проходящих субэпикардиально коронарных артерий и, кроме того, подвергаются наибольшему механическому сдавлению в систолу.
Дата добавления: 2015-09-18; просмотров: 1184;