ГРУППА ВЕЩЕСТВ, ИЗОЛИРУЕМЫХ ИЗ БИОЛОГИЧЕСКОГО МАТЕРИАЛА ЭКСТРАКЦИЕЙ И СОРБЦИЕЙ 2 страница
1. Длительность (8 – 10 рабочих дней) и многостадийность. Большое количество операций, связанных с осаждением белков и фильтрованием, ведёт к значительным потерям искомых веществ (алкалоиды теряются на 25 – 50 %).
2. Сравнительная дороговизна метода (на 1 исследование тратится до 500 мл этанола).
Все это приводит к тому, что классический метод Стаса – Отто теряет своё былое значение и постепенно заменяется более быстрыми, эффективными и экономными методами извлечения подкисленной водой.
В настоящее время метод применяется, главным образом, для исследования гнилостно изменённого биологического материала.
5.2.1.2. Изолирование водой, подкисленной щавелевой кислотой
Идея изолирования подкисленной водой высказывалась разными исследователями. Так, ещё в 1856 году С. Макадам предложил для подкисления воды использовать щавелевую кислоту, в 1861 г. Усляр и Эрдман – соляную, а в 1865 г. Г. Драгендорф – серную. Вплоть до 1941 г. водный метод не получал широкого распространения. В 1942 – 1943 г.г. Степановым А.В. и Швайковой М.Д. был предложен метод изолирования алкалоидов из объектов растительного происхождения водой, подкисленной щавелевой кислотой (ускоренный метод). В 1947 – 49 г.г. этот метод был применён А.А. Васильевой к трупному материалу, после чего он вошёл в практику судебно–химического анализа в отечественных лабораториях.
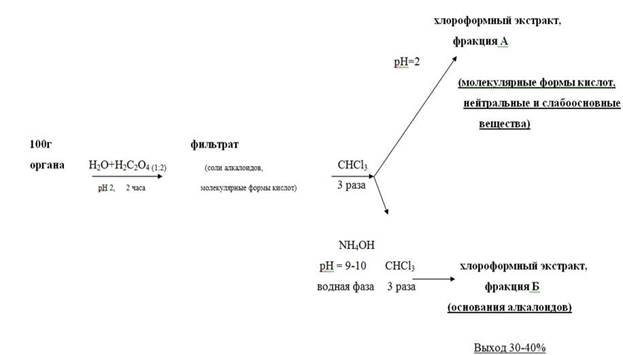
Схема изолирования по методу Васильевой заключается в следующем:
• Настаивание измельчённого объекта с водой, подкисленной щавелевой кислотой до рН = 2 – 3, в течение двух часов. Вода берётся в количестве 1 : 2 по отношению к навеске объекта. Водное извлечение фильтруется.
• Экстрагирование веществ кислого, нейтрального и слабоосновного характера из водного фильтрата хлороформом при рН = 2 (трёхкратная экстракция), отделение органической фазы и концентрирование полученного извлечения упариванием (фракция А, «кислое» извлечение).
Подщелачивание оставшегося после разделения фаз водного слоя раствором аммиака до рН 9 – 10, экстрагирование веществ основного характера трёхкратной экстракцией хлороформом, отделение органической фазы и концентрирование упариванием (фракция Б, «щелочное» извлечение).
По сравнению с изолированием подкисленным спиртом извлечение водой, подкисленной щавелевой кислотой, обладает рядом преимуществ:
1. Быстрота (анализ можно провести в течение одного рабочего дня).
2. Меньшее количество операций, меньшие потери искомых веществ (алкалоиды извлекаются на 30 – 40 %).
3. Экономичность и дешевизна, т.к. дорогой спирт заменён водой.
Недостатком метода является образование стойких эмульсий при экстрагировании веществ из водной фазы хлороформом, особенно при исследовании гнилостного биоматериала, т.к. метод не предусматривает очистки извлечений
Рисунок 2. Схема проведения анализа методом Васильевой
5.2.1.3. Изолирование нейтральным ацетоном
Метод предложен проф. Карташовым В.А. (г. Барнаул) в 1990 году.
Измельченную навеску биологического материала массой 5 г экстрагируют 10 мл ацетона в течение 10 минут, центрифугируют. Полученное извлечение сливают, операцию экстрагирования ацетоном повторяют ещё 2 раза по 5 мл. Извлечения объединяют.
К объединенному извлечению добавляют 20 мл 0,5 М. раствора хлористоводородной кислоты, перемешивают и экстрагируют гексаном дважды. Гексановые извлечения, содержащие примеси, отбрасывают.
Из очищенного водно-ацетонового извлечения (рН=1) эфиром экстрагируют вещества кислотного характера. Далее водно-ацетоновую фазу подщелачивают аммония гидроксидом до рН=9, добавляют 5 г натрия хлорида (высаливающий агент) и экстрагируют вещества основного характера эфиром.
Преимущество метода:
1. Высокий выход ядовитых веществ (до 60-70%). Это достигается за счет использования в качестве экстрагента нейтрального ацетона, который извлекает вещества кислотного и основного характера как в ионизированной, так и в молекулярной формах. Высокий выход веществ позволяет уменьшить массу навески до 5 граммов, что упрощает операции и снижает расход реактивов и времени пробоподготовки.
Недостатки метода:
Ацетон, извлекает из биоматериала большое количество примесей, что требует дополнительной очистки извлечения. К недостаткам метода относится и его многостадийность.
5.2.1.4. Твердофазная экстракция
Лекарственные и наркотические средства, поступающие на исследование, крайне редко являются индивидуальными соединениями. Нередко объектами исследования являются поступающие из незаконного оборота синтетические наркотические средства, которые производятся в подпольных лабораториях. В подавляющем большинстве случаев они имеют в своем составе различные наполнители и добавки (сахар, соли жирных кислот, крахмал, сода, тальк и др.), либо содержат загрязнения или промежуточные и побочные продукты синтеза, содержание же наркотически активного компонента в таких препаратах очень низкое.
При этом разделение, выделение, концентрирование и очистка целевых компонентов традиционными методами (например, жидкостной экстракцией) неэффективны, а иногда просто невозможны. Решение такого рода проблем возможно при применении на стадии пробоподготовки метода твердофазной экстракции, позволяющего осуществлять одновременное разделение, выделение и концентрирование целевых компонентов из биологических жидкостей и их экстрактов, лекарственных и нативных наркотических средств. Такой подход дает возможность получения веществ в чистом виде, что в дальнейшем позволяет проводить идентификацию методами ИК -, УФ – спектроскопии, ТСХ, ГЖХ, ВЭЖХ, методом хромато-масс-спектрометрии.
В основе метода твердофазной экстракции лежит принцип колоночной хроматографии, который основан на специфическом взаимодействии выделяемого из биоматериала компонента с сорбентом, находящимся в небольшом патроне. Патрон – картридж имеет полиэтиленовую оболочку, внутри которой находится сорбент, упакованный между двумя пористыми фильтрами. Патроны могут соединяться друг с другом, представляя более широкие возможности для их использования. Чаще всего для заполнения патронов применяют сорбенты на основе силикагеля и химически модифицированного силикагеля. Для модификации силикагеля используются вещества, содержащие различные функциональные группы (нитрильные, диольные, амино-, карбокси - и сульфогруппы), а также алифатические (С1 – С18) и ароматические (фенильные) группы. Выбор соответствующего типа патрона связан со свойствами определяемого вещества и осуществляется по типу подобия.
Часто в практике ХТА используются отечественные патроны фирмы “Диапак”, заполненные немодифицированным силикагелем (Диапак Силикагель) и силикагелем, модифицированным гексадецильными группами С16 (Диапак С16).
При использовании патронов Диапак для большинства веществ проводят следующие операции.
1. Активация – приведение патрона в рабочее состояние путем промывки этиловым или метиловым спиртом (2-10 мл). Скорость пропускания 2,5 мл/мин.
2. Кондиционирование – пропускание буферного раствора (ацетатного или аммиачного) в зависимости от исследуемых веществ. Объем буферного раствора – 10 мл. Скорость пропускания 2,5 мл/мин .
3. Сорбция – пропускание исследуемого раствора через патрон. Объем пробы обычно 100 мл. Скорость пропускания 2,5 мл/мин.
4. Десорбция – удаление исследуемого вещества с сорбента с помощью воздуха шприцем или вакуумным насосом.
5. Элюирование образца осуществляется реагентом, который применялся на стадии активации. Объем элюента – 50-100 мл.
Высокая эффективность выпускаемых патронов позволяет использовать их для пробоподготовки широкого круга объектов – от лекарственных препаратов сложного состава до “уличных наркотиков” и биологических жидкостей (моча, кровь, сыворотка и т. д.), а также экстрактов биологических жидкостей.
Частные методы изолирования веществ кислотного характера
• изолирование барбитуратов подщёлоченной водой (метод П. Валова);
• метод Е. Грусц-Харди.
5.2.1.5. Изолирование барбитуратов подщелоченной водой (метод П. Валова)
Изолирование барбитуратов подщелоченной водой схематично можно представить следующим образом:
• Настаивание измельчённого объекта с водой, подщелоченной 20% раствором натрия гидроксида до рН = 12 и более в течение 30 мин.
• Очистка водного извлечения путём насыщения натрия вольфраматом, фильтрование раствора.
• В кислой среде (серной кислотой до рН=2), экстрагирование эфиром, концентрирование эфирного извлечения упариванием.
Выход 50% и более
Выход составляет 50% и даже до 90%, в зависимости от вида барбитурата. Метод даёт достаточно чистые извлечения, т.к. включает стадию очистки (осаждение белков натрия вольфраматом), что повышает качество последующего анализа.
Недостатком метода является соосаждение барбитуратов с белками при обработке натрия вольфраматом. В последней модификации метода серная кислота заменена на натрия гидросульфат, что увеличивает выход искомых веществ.
Рисунок 3. Схема проведения анализа методом П. Валова

Измельченный биологический материал растирают с сульфатом аммония, подкисляют раствором хлористоводородной кислоты до рН=2, экстрагируют смесью этилового спирта и хлороформа (1:1). Экстракт отделяют, выпаривают, сухой остаток растворяют в горячей воде и фильтруют. Из фильтрата экстрагируют вещества кислотного характера эфиром.
Частные методы изолирования веществ основного характера
5.2.1.7. Частный метод изолирования алкалоидов водой, подкисленной серной кислотой (по В.Ф. Крамаренко)
Метод был разработан Львовской школой судебных химиков под руководством заведующего кафедрой аналитической и токсикологической химии Львовского медицинского института проф. В.Ф. Крамаренко. В 1956 – 62 г.г. целый ряд работ химиков этой школы был посвящён влиянию рН среды, природы экстрагента и присутствия в водной фазе электролита на эффективность изолирования алкалоидов из биоматериала.
Схематично метод можно представить из следующих этапов:
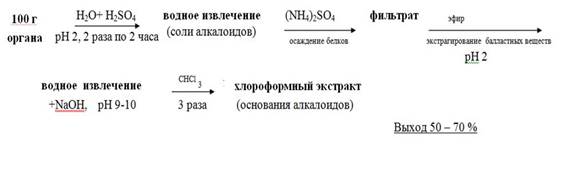
• Настаивание измельчённого объекта с водой, подкисленной 20% раствором кислоты серной до рН =2– 3, в течение двух часов. Вода берётся в количестве 1 : 2 по отношению к навеске объекта. Водное извлечение фильтруется. Операция повторяется двукратно.
• Очистка водного извлечения от белковых соединений путём насыщения его аммония сульфатом, настаивания в течение часа и фильтрования образовавшегося осадка.
• Очистка фильтрата от жиров, смол, пигментов путём экстракции эфиром. Эфирное извлечение отбрасывают.
• Подщелачивание водного извлечения 20% раствором натрия гидроксида и экстрагирование веществ основного характера хлороформом при рН =9–10 (трёхкратная экстракция), отделение органической фазы и концентрирование полученного извлечения упариванием.
Разработанный вначале для алкалоидов, метод применим и для изолирования других азотсодержащих веществ основного характера (синтетических лекарственных средств).
Метод достаточно быстрый. Преимуществом является хорошая очистка извлечений от соэкстрактивных веществ.
Рисунок 4. Схема проведения анализа методом В.Ф.Краморенко
Кроме указанных выше методов изолирования, для отдельных алкалоидов, таких, как жидкие алкалоиды – анабазин, никотин, кониин рекомендуется перегонка алкалоидов с водяным паром с последующим экстрагированием из дистиллята органическим растворителем. Для стрихнина и пахикарпина предложены такие методы, как электрофорез и электродиализ.
5.2.1.8. Исследование биологических жидкостей
1. При исследовании биожидкостей, таких как кровь, моча, плазма, слюна, сыворотка, промывные воды желудка, для изолирования «нелетучих» ядов используют прямую дробную жидкость – жидкостную экстракцию (ЖЖЭ).
Биожидкость подкисляют кислотой хлористоводородной до рН 2-3 и экстрагируют эфиром (фракция А), а затем подщелачивают до рН 9-10 и экстрагируют хлороформом (фракция Б). Во фракции А присутствуют вещества кислого, нейтрального и слабоосновного характера, во фракции Б – вещества основного характера.
ЖЖЭ соответствует второй стадии изолирования ядов из биоматериала в рассмотренных ранее общих методах, поэтому все факторы, определяющие эффективность изолирования на этой стадии, имеют место здесь.
Для некоторых веществ основного характера (морфин) при изолировании его из биожидкостей предварительно проводят кислотный гидролиз, чтобы разрушить его комплекс с глюкуроновой кислотой, в виде которого он находится в жидкостях. При прямой ЖЖЭ совместно с ядом из биожидкостей могут экстрагироваться сопутствующие вещества, что заставляет в дальнейшем прибегать к различным методам очистки.
В последнее время для изолирования ядов из биожидкостей применяется сорбция их на синтетических смолах, модифицированных силикагелях и активированном угле. Вещества сорбируются на твердом носителе, а потом элюируются из него органическим растворителем (по фракциям). Метод позволяет не проводить дополнительную обработку пробы и изолирование, дает возможность одновременно сконцентрировать вещество и провести очистку. Это приводит к повышению чувствительности метода. Однако, при неизвестном яде сорбция не всегда может быть использована ввиду опасности его потери из-за недостаточной сорбции.
5.3. АНАЛИТИЧЕСКИЙ СКРИНИНГ ЛЕКАРСТВЕННЫХ ВЕЩЕСТВ, ИМЕЮЩИХ ТОКСИКОЛОГИЧЕСКОЕ ЗНАЧЕНИЕ
1. Необходимость разработки аналитических скрининговых методов анализа вызвана многообразием лекарственных соединений, которые при определенных обстоятельствах могут стать источником отравлений. Круг этих веществ постоянно расширяется по мере синтеза новых соединений.
Рост числа отравлений лекарственными препаратами связан с самолечением, в результате чего имеют место случаи передозировки лекарств, проявление их побочного действия и развитие идиосинкразии.
Большую долю в общем числе отравлений лекарственными веществами составляют детские отравления. Определенный вклад вносят отравления, связанные с наркоманией и токсикоманией.
Клиническая диагностика острых отравлений лекарственными средствами затруднена из-за нехарактерных симптомов. Особенно трудно идентифицируются комбинированные и детские отравления, где природа яда чаще неизвестна. В связи с этим возникает необходимость в разработке «скрининговых» методов анализа, которые позволяли бы за короткое время выбрать из большого числа потенциальных ядов одно или несколько веществ, послуживших причиной отравления. «Скрининг» (англ.) – отсеивание, просеивание. Таким образом, СКРИНИНГ – это научно обоснованная система поиска неизвестного яда, когда в процессе последовательных операций поэтапно отсеиваются (или определяются) отдельные группы веществ или индивидуальные вещества.
Аналитический скрининг наиболее удобен и эффективен в случае аналитической диагностики комбинированных отравлений, а также при ненаправленном анализе, когда неизвестна природа определяемого вещества. Применение аналитического скрининга позволяет систематизировать исследование и направить его в нужное русло, сократив тем самым время анализа и материальные затраты на его проведение.
К аналитическим скрининговым методам предъявляются определенные требования:
-универсальность (возможность подвергнуть исследованию большое количество веществ);
- достаточная специфичность (чаще групповая);
- высокая чувствительность (до десятых долей мкг);
- экспрессность (возможность выполнения серийных анализов);
- точность (отклонение до 10%) и воспроизводимость;
- простота и доступность;
- лабильность (возможность оптимизации и модернизации за счет введения новых соединений, материалов, реагентов и оборудования в существующую систему скрининга);
- возможность сочетания с другими методами анализа.
Перечисленным требованиям удовлетворяют, в основном, современные физико-химические методы анализа:
1. Хроматографические.
2. Спектроскопические.
3. Иммунохимические.
Эти методы могут быть использованы в системе как общего, так и частного скрининга.
Общий скрининг – предусматривает, в основном, химическое исследование веществ, отличающихся по своему строению, и принадлежащих к различным фармакологическим группам. В основном здесь применяется групповая идентификация (например, группа производных барбитуровой кислоты, фенотиазина и т.п.)
Частный скрининг – направлен на исследование веществ внутри группы и идентификацию ее отдельных представителей. Примером может служить хроматографическое исследование на производные барбитуровой кислоты для идентификации конкретного вещества из этой группы.
5.3.2.Хроматографические скрининговые методы
Хроматографию можно рассматривать как метод разделения веществ, в основе которого лежит разница в коэффициентах распределения этих веществ между неподвижной и подвижной фазами.
В настоящее время в качестве методов хроматографического скрининга наибольшее применение находят:
1. Тонкослойная хроматография (ТСХ).
2. Высокоэффективная тонкослойная хроматография (ВЭТСХ).
3. Газожидкостная хроматография (ГЖХ).
4. Высокоэффективная жидкостная хроматография (ВЭЖХ).
5.3.2.1.Тонкослойная хроматография (ТСХ)
В ТСХ роль неподвижной фазы выполняет тонкий фиксированный слой сорбента (0,1-0,5 мм), содержащий определенное количество воды, нанесенный на пластинку из стекла (алюминиевой фольги, полимера). В качестве сорбентов чаще используется силикагель и окись алюминия. Для лучшего связывания сорбента в него добавляют связующий компонент – гипс, крахмал и др. Роль подвижной фазы выполняет индивидуальный растворитель или смесь растворителей, называемые «хроматорафической системой».
В процессе хроматографирования по мере подъема растворителя вверх по пластине за счет капиллярных сил происходит разделение смеси веществ согласно их коэффициентам распределения между подвижной и неподвижной фазами.
Детектирование (обнаружение) веществ на хроматограмме проводят следующим образом:
- визуально, если вещество имеет собственную окраску (дротаверин, клозапин, бензофеноны и др.),
- в УФ-свете – для веществ, обладающих флуоресценцией (свечением) или способных поглощать УФ-излучение (темные пятна на флуоресцирующем фоне),
- с помощью хромогенных реакций: бесцветные вещества переводят в окрашенные соединения, обрабатывая хроматографическую пластинку соответствующими реактивами.
Идентификацию веществ на хроматограммах проводят по коэффициенту Rf (скорость фракционирования).
Rf – это величина, численно равная отношению длины пробега анализируемого вещества к длине пробега растворителя:
Rf = Нв-ва / Нраств.
Rf является константой вещества в заданных хроматографических условиях при их строгом соблюдении. На практике соблюдение всех условий затруднено. Величина Rf может колебаться в зависимости от целого ряда факторов:
- техники выполнения работы,
- качества и активности сорбента,
- толщина слоя сорбента,
- чистоты растворителей,
- степени насыщения камеры парами растворителей,
- температуры,
- присутствия балластных веществ и проч.
Поэтому иногда принято пользоваться не абсолютным, а относительным значением Rf, которое обозначается как Rst.
Относительный коэффициент Rst. численно равен отношению абсолютной величины Rf исследуемого вещества к Rf метчика:
Rst = Rf в-ва / Rf метчика
В качестве метчика используется вещество, принятое за стандарт. Относительное значение Rf более воспроизводимо, т.к. с изменением хроматографических условий меняются значения Rf исследуемого веществ и вещества-стандарта, а их соотношение остается достаточно стабильным. При совпадении величин Rf исследуемого веществ и вещества-стандарта можно предположить их идентичность. В условиях скрининга Rf исследуемого вещества сравнивают с табличными данными, полученными в аналогичных условиях
Метод ТСХ широко распространен благодаря своей доступности и простоте выполнения, и в то же время высокой эффективности, чувствительности, экспрессности, достаточной специфичности.
ТСХ применяется в системе общего и частного скрининга и разработана для многих лекарственных веществ, имеющих токсикологичское значение.
В общем скрининге используется множество систем растворителей, из них можно выделить некоторые:
1. Для веществ кислого, нейтрального и слабоосновного характера, извлекаемых органическим растворителем из кислого водного раствора – хлороформ:ацетон (9:1).
2. Для веществ основного характера, извлекаемых органическим растворителем из щелочного водного раствора: диоксан:хлороформ:ацетон:25% раствор аммиака (47,5:45:5:2,5), ацетон:хлорофром:25% раствор аммиака (245:12:1).
3. При анализе наркотических и других одурманивающих веществ, при экспресс-анализе острых отравлений используют универсальную систему толуол:ацетон:этанол:25% раствор аммиака (45:45:7,5:2,5).
Для детектирования веществ на хроматографической пластинке разработана схема последовательного их проявления.
Для веществ кислого, нейтрального и слабоосновного характера:
- УФ-облучение,
- дифенилкарбазон в хлороформе и раствор сульфата ртути: проявляются производные барбитуровой кислоты;
- раствор хлорида железа трехвалентного: проявляются производные пиразолона, салициловой кислоты, фенотиазина;
- реакив Драгендорфа и 0,5 М серная кислота: проявляются азотсодержащие вещества слабоосновного характера.
Для веществ основного характера:
- УФ-облучение,
- раствор хлорида железа трехвалентного: проявляются производные пиразолона, фенотиазина;
- раствор натрия нитрата в кислоте хлорной: проявляются производные фенотиазина, тиоксантены;
- реакив Драгендорфа и 0,5 М серная кислота: проявляются азотсодержащие вещества основного характера.
Отдельная пластинка: реактив Марки (концентрированная кислота серная, содержащая 10% формалина): проявляются, в частности алкалоиды группы опия, дифенгидрамин, некоторые производные фенотиазина и др.
При обнаружении веществ в общих системах переходят к исследованию в частных системах растворителей.
Например:
Для барбитуратов – хлороформ:н-бутанол:25% раствор аммиака (70:40:5).
Для алкалоидов опия – этилацетат:метанол:25% раствор аммиака (17:2:1).
Разработанная методика проста, универсальна, может быть использована в клинико-диагностических лабораториях и Бюро судебно-медицинской экспертизы.
С помощью ТСХ можно определять вещества в количествах от 10-9 до 10-6 г, ошибка определения компонентов – 5-10 %.
Воспроизводимость результатов исследования методом ТСХ достигается проведением их при следующих условиях:
1. Инструментальные условия проведения эксперимента, которые включают конструкцию используемой хроматографической камеры; способ ее герметизации; условия насыщения камеры парами растворителя и т.д.
2. Свойства хроматографической системы, которые включают тип и способ химической обработки использованного сорбента; величину его зернения; толщину слоя, а также тип подложки, на которую он нанесен; вид и количество внесенных в адсорбент вспомогательных веществ, таких как связующие компоненты и флуоресцирующие вещества; метод активации сорбента, например, выдерживание при повышенной температуре; способ обработки пластинки импрегнирующими буферами, щелочами или кислотами, а также веществами, модифицирующими ее свойства.
3. 3. Методические подходы к нанесению пробы и проведению хроматографирования, которые предусматривают способ нанесения образца на пластинку и использованное при этом устройство; размер начальной зоны хроматографирования; полярность растворителя, использованного для нанесения образца и его количество; продолжительность исследования и величину пробега растворителя; его состав и чистоту; температуру и влажность окружающей среды в момент проведения исследования.
Представление результатов исследований. В экспертном заключении полученные рассматриваемым методом результаты должны содержать подробное описание условий проведения эксперимента: хроматографические пластины (адсорбент, наличие или отсутствие индикатора в его составе, использованное связующее вещество, тип подложки для адсорбента, фирма – изготовитель, а также проведение специальной обработки пластин перед исследованием, например, высушивание при повышенной температуре или импрегнирование щелочью или буфером). Схема обработки хроматографических пластин должна содержать описание использованных реактивов с их полным качественным и количественным составом, последовательности проведения этапов обработки, их интенсивности и продолжительности. Особо отмечается интенсивность и окраска хроматографических зон исследуемых веществ после обработки каждым реактивом, а также величина их Rf или RRf. При проведении подтверждающих исследований на конкретное вещество допускается указание о том, что в данных конкретных условиях величина Rf и окраска хроматографической зоны исследуемого вещества использованными реактивами совпадает с величиной Rf и окраской стандартного раствора - метчика. В заключении приводятся также данные о пределе обнаружения метода.
5.3.2.2. Высокоэффективная тонкослойная хроматография (ВЭТСХ)
ВЭТСХ появилась в начале 70-х г.г. прошлого столетия как новое направление в ТСХ. Имеет ряд преимуществ по сравнению с ТСХ:
ü более высокая эффективность (за 1 прием можно разделить около 40 веществ),
ü высокая чувствительность и экспрессность.
Высокая эффективность данного метода достигается за счет применения высокодисперсного сорбента, а более высокая чувствительность и экспрессность – более тонкого слоя сорбента (10-15 нм).
В качестве сорбентов применяются силикагель, окись алюминия, целлюлоза, метилцеллюлоза, полиамид, инактивированный силикагель и др.
Методы хроматографирования существенно не отличаются от ТСХ. Методы детектирования идентичны, только более чувствительны. Возможно проведение количественного анализа с применением метода сканирующей денситометрии, дающей точные результаты и не требующей элюирования веществ с пластинки.
ВЭТСХ, так же как и ТСХ, сочетают с другими физико-химическими методами анализа – спектральными, ГЖХ, ВЭЖХ, что повышает надежность исследования.
5.3.2.3. Газожидкостная хроматография (ГЖХ)
ГЖХ нашла свое применение в анализе лекарственных веществ в качестве скринингового метода благодаря своей универсальности. Необходимым условием является переведение исследуемого вещества в летучее состояние. Метод используется при анализе барбитуратов, алкалоидов и других лекарственных веществ в качестве общего и частного скринингового.
Идентификация веществ проводится по относительным временам удерживания (или индексам удерживания Ковача). Количественное определение – по высоте (площади) пиков исследуемого вещества и вещества, применяемого в качестве внутреннего стандарта.
Достоверность исследований повышается при использовании не менее 2 колонок, обладающих различной полярностью. Используемые детекторы – пламенно-ионизационный, масс-селективный.
Дата добавления: 2015-08-01; просмотров: 4916;