СОВРЕМЕННЫЕ МЕТОДЫ АНАЛИЗА МЕТАЛЛОВ, ИСПОЛЬЗУЕМЕ В АНАЛИТИЧЕСКОЙ И ТОКСИКОЛОГИЧЕСКОЙ ХИМИИ (КРАТКИЙ ОБЗОР) 1 страница
В настоящее время для целей аналитической и токсикологической химии при исследовании на металлы используются как химические, так и физико–химические (инструментальные) методы анализа.
Эти методы дополняют друг друга. К достоинствам химических методов можно отнести простоту исполнения, малую стоимость реактивов и оборудования, наглядность получаемых результатов. Химический метод анализа может быть поставлен даже в слабо оснащённой лаборатории.
Физико–химические методы стали активно внедряться в практику аналитической химии приблизительно с 30–х годов 20 века, а начиная с 60–х годов в повседневную практику передовых химических лабораторий внедряется и электронно–вычислительная техника.
В результате стремительного развития инструментального анализа стало возможным регистрировать присутствие соединений, содержание которых не превышает 10–12 г.
Наиболее часто в анализе металлов используются методы атомной спектроскопии. Среди них следует выделить традиционные:
1) атомно–эмисcионную спектроскопию, которой исследуют линейчатые спектры возбуждённых (тем или иным способом) атомов, определяя при-роду и количество отдельных элементов;
2) атомно–абсорбционную спектроскопию, с помощью которой измеряют резонансное поглощение излучения определённой длины волны.
В основе последнего метода лежит закон излучения Кирхгофа, согласно которому элемент поглощает излучение той же длины волны, которое он испускает в возбуждённом состоянии.
В каждом из методов проба переводится в атомарное состояние с помощью различных источников (пламя, дуга, искра, лазер, лампа с полым катодом, печь с графитовыми стержнями).
В атомно–эмиссионной спектроскопии атомы, находясь в возбуждённом состоянии, излучают свет разной длины волны. Для выделения характеристического излучения используют разные оптические приспособления, основанные на явлениях дифракции и интерференции. А также преломления и фокусировки света.
Разложенный свет содержит собственные окрашенные возбуждённые компоненты – фон спектра, на котором чётко выделяются более яркие линии возбуждённых атомов анализируемого вещества.
Если ещё совсем недавно спектры регистрировались на бумаге, обработанной светочувствительными красителями, на которой нанесены положения линий некоторых известных элементов, то внедрение компьютеров позволяет использовать для идентификации вещества не только несколько отдельных характеристических линий, а весь спектр, разрешённый с точностью до нанометра.
По сути явление атомизации пробы и возбуждения образовавшихся атомов, в эмиссионной спектроскопии, можно рассматривать как единый процесс, протекающий в одном устройстве прибора.
В отличие от этого вида спектроскопии в атомно–абсорбционной спектроскопии атомизированная проба освещается от внешнего источника световым потоком. При прохождении пробы интенсивность светового потока уменьшается, так как атомы определяемого элемента поглощают свет определённой длины волны. Так как измерить изменение интенсивности белого света довольно сложно, используется монохроматор. Однако, в настоящее время используется другое техническое решение проблемы: свет должен исходить от возбуждённых атомов того самого элемента, который необходимо обнаружить в пробе. Для этой цели используются лампы с полым катодом. Внутри катодной полости, покрытой металлом или сплавом требуемого элемента, создаётся высоковольтный разряд, и атомы материала катода начинают излучать характеристические фотоны. Когда излучение пропускают через газообразную (атомизированную) пробу, его интенсивность уменьшается, и по разности в интенсивностях определяют содержание элемента в пробе. Так как для каждого элемента требуется своя катодная лампа, этот метод рентабелен лишь для серийных анализов. В современных приборах часто применяют многоэлементные катодные лампы, хотя, для возможных комбинаций элементов существуют известные ограничения.
Оба метода высокочувствительны и специфичны. Предел обнаружения лежит в интервале 10–4 – 10–12 г в зависимости от определяемого элемента и используемой разновидности метода. (Разница в методах определяется способом атомизации и технической конструкцией прибора). Методы могут использоваться как для качественного, так и для количественного анализа. В количественных определениях применяют метод градуировочного графика и метод добавок. Следует отметить, что воспроизводимость результатов в количественных расчётах в методе атомно–абсорбционной спектроскопии выше, чем атомно–эмиссионной.
Ещё более чувствительным и универсальным является активационный анализ. В настоящее время известно несколько методов: фотонный активационный анализ, активационный анализ с помощью заряженных частиц и нейтронно–активационный анализ.
Наиболее часто используется нейтронно–активационный анализ, который требует для своего проведения сложного оборудования, однако принцип, лежащий в основе этого метода прост. Большинство химических элементов в обычных условиях не являются радиоактивными, но после облучения становятся радиоактивными. Для облучения используют нейтральные частицы – нейтроны атомного реактора, либо другие радиоактивные нейтронные генераторы или радиобериллиевые излучатели. Два последних источника характеризуются значительно более слабым нейтроннным потоком, чем реакторы.
Ядра стабильного элемента, взаимодействуя с нейтронами, превращаются в ядра радиоактивного элемента и испускают излучение с характерной энергией. Регистрируя это излучение, можно установить, какому радиоактивному элементу оно принадлежит. Измеряют, в основном, g–излучение, электромагнитное по своей природе и имеющее аналогию с видимым светом, но отличающимся от него более высокой энергией.
В g–спектре легко выделить отдельные пики, различающиеся между собой по энергии излучения; каждый пик может указывать на присутствие определённого элемента. Аналитические определения, как правило, проводят с помощью стандартов, которые облучают вместе с образцом. Измерение интенсивности пиков позволяет получить достоверные сведения о концентрации искомых элементов.
Лишь очень немногие методы анализа допускают возможность исследования пробы без какой–либо предварительной подготовки, в исходном состоянии. Тем более, что это утверждение в большей мере справедливо для ограниченно компонентных и однородных образцов. Такой же сложный объект исследования как биологический материал, содержащий 65 элементов, из которых известна роль только половины, представляет определённые, порой очень большие трудности в анализе данными методами и нуждается в тщательной предварительной подготовке проб.
Однако, если технические затруднения в конечном итоге преодолимы, то экономическая проблема, связанная с дороговизной и рентабельностью оборудования, используемого в методах, делает оснащение этими приборами большинства химико–токсикологическихи криминалистических лабораторий невозможным.
В связи с этим, некоторый интерес представляют электрохимические методы анализа, стоимость оборудования в которых значительно ниже. Так например, вольтамперометрия (полярография) пригодна для определения почти всех неорганических катионов, а её разновидность – инверсионная вольтамперометрия (инверсионная полярография) к тому же очень чувствительна (10–9 – 10–10 моль/л). В основе этого метода лежит концентрирование определяемого вещества на поверхности или в массе электрода с последующей регистрацией анодной вольтамперной кривой. Таким образом, весь процесс состоит из двух стадий:
1) накопление определяемого элемента на электроде (электролиз);
2) получение вольтамперограммы (развёртка потенциала).
Для проведения электролиза широко применяют различные типы рабочих электродов: ртутноплёночные различной формы, так как для повышения чувствительности метода необходимо уменьшить объём ртути; платиновые; золотые; стеклоуглеродные; стеклографитовые и другие.
Ртутно–плёночный электрод на серебряной подложке представляет собой тонкую плёнку ртути (20 – 100 мкм), нанесённую на отшлифованную серебряную проволоку или путём погружения её в чистую ртуть, или путём электролиза. Серебряная подложка крепится в инертном материале – стекле, фторопласте или полиэтилене. Поверхностная плёнка ртути тщательно растирается (калькой, фильтром) по серебру для предотвращения контакта серебра с рабочим раствором. Стеклоуглеродный, стеклографитовый, золотой и другие электроды представляют собой стеклянные полые стержни, в которые запакованы под вакуумом медная проволока с прикреплённым серебряным стержнем, выполняющим функции проводника и служащим для закрепления электрода в приборе, а также соответствующей рабочей поверхностью, которая помещается в рабочий раствор.
Электродом сравнения служит хлорсеребряный. Хлорсеребряный электрод изготавливают путём нанесения хлористого серебра разными способами на серебряную поволоку. При электрохимическом способе очищенную серебряную спираль погружают в 0,01-0,1 моль/л раствор кислоты хлороводородной и присоединяют её к положительному полюсу сухой батареи на 1,5 В. К отрицательному полюсу через реостат и миллиамперметр присоединяют погружённую в кислоту платиновую проволочку. С помощью реостата регулируют силу тока (1 - 10) мА и пропускают ток в течение нескольких минут, чтобы на поверхности серебряного анода образовался тонкий слой серебра хлорида. После электролиза электрод помещают во фторопластовую трубочку с раствором калия хлорида соответствующей концентрации и отделяют от анализируемого раствора пористой пробкой (из стекла, керамики и т.д.).
На рабочий электрод подают потенциал на несколько десятых долей вольта отрицательнее полуволны определяемого иона (см. полярографию) и катионы металла начинают восстанавливаться на поверхности электрода до свободного металла. Обычно в процессе концентрирования выделяется только часть определяемого вещества, поэтому для получения количественных результатов необходимо не только контролировать потенциал, приложенный к рабочему электроду, но и тщательно воспроизводить размер электрода, продолжительность электролиза и скорость перемешивания как анализируемого, так и стандартного раствора, применяемого для калибровки.
В конечном итоге, чувствительность вольтамперометрического метода, используемого на завершающей стадии анализа, определяется продолжительностью стадии электролиза. По окончании электролиза перемешивание прекращают и дают раствору успокоиться в течение примерно 30 секунд. Затем потенциал, приложенный к рабочему электроду, линейно с заданной скоростью изменяют (разворачивают) в анодном (положительном) направлении и наблюдаемое изменение тока, проходящего через электролитическую ячейку, регистрируют как функцию наложенного потенциала. Ток пика вещества (или высота пика) после поправки на остаточный ток прямо пропорционален его концентрации. Концентрацию вещества находят по калибровочному графику, построенному по стандартным растворам или методом внутреннего стандарта.
Следует отметить перспективность использования таких методов как хроматография (тонкослойная, бумажная), электрофорез, принципы которых должны быть вам хорошо знакомы. Основное назначение этих методов – разделение веществ. Чувствительность качественного анализа электрофорезом и хроматографией определяется чувствительностью химических реакций обнаружения, которые используются для проявления электрофореграмм и хроматограмм. С целью повышения чувствительности анализа указанными методами рекомендуется проявлять металлы люминесцентными реакциями, основанными на способности многих катионов изменять характер люминесценции различных органических реагентов. Кроме того, меняя рН среды с помощью буферных растворов, можно одним и тем же реактивом провести обнаружение катионов и анионов в смеси. Например, 8–оксихиноляты металлов флюоресцируют при различных значениях рН.
4. ГРУППА ВЕЩЕСТВ, ИЗОЛИРУЕМЫХ ДИСТИЛЛЯЦИЕЙ («ЛЕТУЧИЕ ЯДЫ»)
В данной главе дана общая характеристика группы «летучих ядов»; основы перегонки с водяным паром (для простых и азеотропных смесей). Определены объекты судебно-химического исследования; основы пробоподготовки; необходимая аппаратура и техника перегонки для проведения анализа на группу «летучих ядов». Рассмотрены современные методы изолирования, их характеристика, дана сравнительная оценка методам (рассмотрена дистилляция с водяным паром, микроперегонка и микродиффузия).
Определено токсикологическое значение некоторых летучих ядов. Показано использование химических реакций при обнаружении «летучих ядов».
Отдельно рассмотрено обнаружение цианидов методом микродиффузии. Показано количественное определение цианидов. Рассмотрено обнаружение алкилгалогенидов, альдегидов и кетонов, фенола, уксусной кислоты.
Отдельно рассмотрены спирты, токсикокинетика спиртов, распределение в организме, биотрансформация, экскреция. Определены объекты исследования при проведении анализа на спирты. Рассмотрены правила отбора проб у живых лиц, трупного материала. Показаны химические свойства спиртов, химические реакции для обнаружения спиртов в дистилляте (метанола, этанола, изоамилового спирта, пропилового, бутилового спиртов и этиленгликоля).
Особое внимание уделяется этиловому спирту. Показаны свойства, механизм действия на организм человека; его токсичность. Определены проблемы и распространенность алкоголизма, экспертиза алкогольного опьянения, клиника отравлений этиловым спиртом. Выделены методы анализа, применяемые в определении наркотического опьянения (качественно-количественные). Показаны предварительные качественные пробы на этиловый алкоголь при исследовании выдыхаемого воздуха и биологических жидкостей (проба Рапопорта А.М., индикаторные трубки Мохова – Шинкаренко).
Показана необходимость количественного определения спиртов (химические и современные биохимические методы исследования; газохроматографический метод исследования этилового спирта).
4.1. ОБЩАЯ ХАРАКТЕРИСТИКА ГРУППЫ
В химико-токсикологическом анализе деление токсикологически важных веществ на группы основано на способах их изолирования из исследуемого объекта. Таких групп насчитывается шесть, причем три из них подлежат обязательному судебно-химическому исследованию при проведении полного (общего) судебно-химического анализа.
Одной из групп токсикологически важных веществ, подлежащих обязательному исследованию, являются «летучие яды», или вещества, изолируемые дистилляцией. Все они изолируются из биологического объекта одним из наиболее старых и широко используемых методов дистилляцией - перегонкой с водяным паром.
В группу «летучих ядов» входят вещества, различные по своей химической природе:
1. Синильная кислота HCN имеет собственную низкую температуру кипения + 26,5°С и занимает первое место по своей летучести с водяным паром.
2. Алкилгалогениды:
СНС13
(хлороформ)
Cl3C-CH(OH)2
(хлоралгидрат)
ССl4
(четыреххлористый углерод)
C2H4CI2
(дихлорэтан)
C2Cl6
(гексахлорэтан)
- Альдегиды и кетоны алифатического ряда:
СН2О (формальдегид)
СНз-СО-СНз (ацетон)
4. Алканолы (спирты):
СНзОН (метанол)
С2Н5ОН (этанол)
С3Н7ОН (пропанол)
С4Н9ОН (бутанол)
С5Н 11ОН (пентанол)
диолы СН2 ОН -СН2 ОН (этиленгликоль)
5. Сложные эфиры алифатического ряда:
CH3COOC2H5 (амилацетат)
6. Карбоновые кислоты алифатического ряда:
CH3COOH (кислота уксусная)
CH3-СНOH-СООН (кислота молочная или альфа-оксипропионовая)
7. Сероуглерод CS2
8. Элементоopгaнические соединения жирного ряда:
(C2H5)4Pb (тетраэтилсвинец)
9. Ароматические углеводороды:
С6Н6 (бензол)
CH3-С6Н5 (толуол)
ксилолы (содержат два радикала -СНз в бензольном кольце в различных положениях)
10. Нитро- и аминопроизводные ароматического ряда:
С6Н5NО2 (нитробензол)
С6Н5NH2 (анилин)
11. Оксипроизводные ароматического ряда:
С6Н5ОН (фенол)
крезолы
кислота салициловая (о-оксибензойная)
12. Фосфор и первые продукты его окисления и восстановления
Н3РО2 (кислота фосфорноватистая)
Н3РО3 (кислота фосфористая)
PH3 (фосфин)
ФОСы (эфиры фосфорных кислот)
13. Жидкие алкалоиды:
кониин
никотин
анабазин
Из перечисленных соединений согласно действующего до настоящего времени приказа Минздрава СССР №1021 от 25.12.73 г., в обязательный круг химико - токсикологического исследования при проведении общего анализа включены:
1. Кислота синильная.
2. Алкилгалогениды: хлороформ, дихлорэтан.
3. Альдегиды: формальдегид.
4. Алканолы: метанол, этанол, пропанол, бутанол, пентанол, изоамиловый спирт.
5. Оксипроизводные ароматического ряда: фенол, крезолы.
По физическим свойствам «летучие» яды, в основном, представляют собой летучие жидкости (за исключением таких твердых веществ, как хлоралгидрат, фенол, салициловая кислота, фосфорорганические соединения).
Способность химических соединений перегоняться с водяным паром зависит от их физических свойств. С водяным паром перегоняются некоторые жидкости, практически не смешивающиеся или ограничено смешивающиеся с водой, азеотропные смеси. Известны также вещества (метанол, ацетон, уксусная кислота, этиленгликоль и др.), которые смешиваются с водой и перегоняются с водяным паром, но не образуют азеотропных смесей.
При перегонке смесей органических веществ большое значение имеет их взаимная растворимость. При этом возможны 3 случая:
1 Жидкости взаимно не растворимы, т.е. образуют двухфазную систему. При перегонке с водяным паром одной из фаз является вода.
2. Жидкости ограниченно растворимы друг в друге, т.е. двухфазная система образуется только при определенных соотношениях компонентов. Такую систему образуют с водой толуол, нитробензол, дихлорэтан, тетраэтилсвинец и др.
3. Компоненты смешиваются в любых соотношениях, т.е. вещества растворимы в воде, образуется однофазная система. С водой такую систему образуют метанол, ацетон, формальдегид, этиленгликоль, уксусная кислота.
В случае образования двухфазной системы (жидкости, не растворимые или ограниченно растворимые в воде) при нагревании смеси давление пара каждой жидкости будет таким же, как и давление ее пара в чистом виде, независимо от наличия другой жидкости. Каждая жидкость в смеси будет вести себя так, как будто отсутствует другая жидкость.
В основе перегонки взаимонерастворимых веществ с водяным паром лежит закон Дальтона.
Согласно этому закону общее давление паров смеси (упругость) равно сумме парциальных давлений (упругостей) ее компонентов при данной температуре.
Р общее = Р воды +Р вещества
При нагревании компоненты смеси увеличивают упругость своих паров независимо друг от друга. Когда общее давление достигнет атмосферного и превысит его на незначительную величину смесь закипает и начинает перегоняться, при этом температура кипения смеси ниже температур кипени каждого из её компонентов в чистом виде за счет сложения их парциальные давлений.
Поскольку одним из компонентов является вода, то вещества буду перегоняться при t°<1000C. Особенно удобна дистилляция с водяным паром в тех случаях, когда изолируемое вещество имеет очень высокую температуру кипения или же разлагается при своей температуре кипения.
Так, для того, чтобы перегонять анилин в чистом виде, требуется нагреть его до температуры кипения, равной 184°С, в смеси же с водой он перегоняется при температуре 75°С.
Такое токсичное вещество, как тетраэтилсвинец, разлагается при своей температуре кипения, равной 200°С.
Кроме того, при проведении судебно-химического исследования сильный нагрев нежелателен, т.к. при высокой температуре может произойти подгорание органических веществ исследуемого объекта и образование следовых количеств синильной кислоты, что приведёт к ложноположительным результатам анализа.
Таким образом, при дистилляции с водяным паром понижается температура кипения перегоняемых соединений и устраняется опасность их термического разложения.
Для многих органических веществ способность перегоняться с водяным может быть объяснена образованием с водой азеотропных (нераздельнокипящих) смесей, состав которых не меняется при перегонке (например, 96% этанола и 4% воды).
Азеотропными называются смеси, у которых пар, находящийся в равновесии с жидкостью, обладает теми же свойствами, что и сама жидкая смесь. Они перегоняются при постоянной температуре и не могут быть разделены простой или фракционной перегонкой.
Из веществ, летучих с водяным паром и представляющих токсикологический интерес азеотропные смеси образуют: алкилгалогениды (хлороформ, ССl4)
§ этиловый и изоамиловый спирты
§ фенол, анилин и др.
В случае образования однофазной системы (жидкости растворимы в воде), если индивидуальная температура кипения вещества низкая (ацетон, метиловый спирт), то оно перегоняется быстро и полностью.
При высокой t°кип обычно полноты отгонки не достигается, при этом приходится использовать селективные переносчики, чтобы образовалась низкокипящая смесь. Так, при перегонке этиленгликоля с водяным паром в качестве селективного переносчика используют бензол, а для уксусной кислоты – гептан. При этом, если температура кипения этиленгликоля составляет 197°С, то смесь этиленгликоль-вода-бензол перегоняется при 118° С. Для уксусной кислоты соответственно 118° С и 80° С.
Оценка метода: очень простой, быстрый, экономичный, не требует специальной аппаратуры. Анализируемые вещества изолируются в чистом виде, только сильно разбавлены водой, поэтому перегонку с водяным паром можно рассматривать не только как метод изолирования, но и как метод очистки.
Объекты судебно-химического исследования. Пробоподготовка
В качестве объектов судебно-химического исследования с целью обнаружения «летучих ядов» на экспертизу обычно направляются внутренние органы трупа, кровь, моча. При подозрении на отравление хлорорганическими веществами дополнительно направляется сальник и 1/3 головного мозга, метанолом - 1/3 головного мозга, этанолом - кровь из крупных вен, моча, мышечная ткань.
Объекты помещают в банки, которые герметично закрывают и опечатывают и немедленно пересылают в лабораторию для исследования. При подозрении на отравление этанолом задержка с транспортировкой материала на 5-10 суток может служить причиной недостоверных результатов его количественного определение.
Методика: Объект в количестве 100 г тщательно измельчают, смешивают с водой до густой кашицы, помещают в круглодонную колбу таким образом, чтобы она заполнилась не более чем на 1/3 её объема, подкисляют кислотой щавелевой или виннокаменной до рН 2-3 и подвергают перегонке.
Подкисление объекта органической кислотой проводят с той целью, чтобы превратить нелетучие соли синильной кислоты (цианиды калия, натрия), в виде которых она находится в биологическом объекте, в свободную HCN, являющуюся легко летучим соединением.
NaCN-------->HCN
В данном случае нельзя воспользоваться сильными минеральными кислотами, т.к. это привело бы:
1) к разрушению молекулы HCN (гидролиз), что приведет к eё потере и недооткрытию.
2) к переоткрытию фенола в результате гидролиза его сернокислых эфиров, (являющихся нормальной частью биологического материала).
4.2. СОВРЕМЕННЫЕ МЕТОДЫ ИЗОЛИРОВАНИЯ «ЛЕТУЧИХ ЯДОВ»
Аппаратура и техника перегонки
Дистилляция с водяным паром проводится в специальном приборе, который состоит из трех основных частей: герметично соединенных друг с другом:
1. Парообразователь
2. Колба с исследуемым объектом (помещается на водяную баню)
3. Холодильник с приемником
Сборку установки начинают со стороны приемника, в последнюю очередь к колбе с исследуемым объектом присоединяют заранее нагретый парообразователь (разборка ведется в обратном порядке). Водяную баню, в которой стоит колба с объектом, также нагревают, чтобы избежать конденсации водяного пара.
Дистилляцию проводят медленно, так, чтобы можно было считать капли отгона
Собирают 2-3 дистиллята. Первый в количестве 1-3 мл собирают в приемник с 5% раствором натрия гидроксида для улавливания легколетучей синильной кислоты и превращения ее в нелетучий цианид натрия. При этом алонж холодильника должен быть погружен в раствор NaOH, чтобы избежать потерь летучей HCN. Весь первый дистиллят используют для обнаружения синильной кислоты.
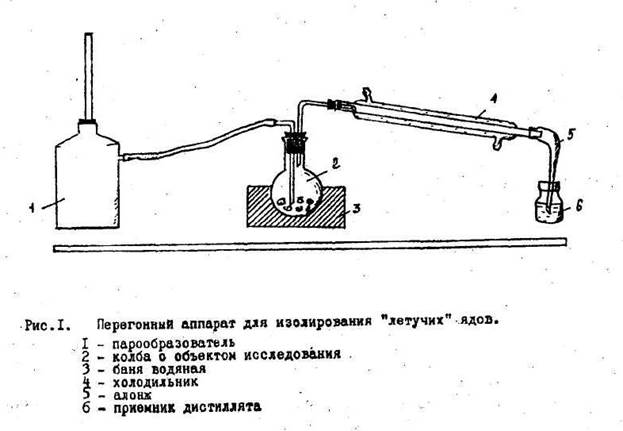
Второй (и третий) дистиллят собирают в пустой, чистый приемник в количестве 20-30 мл и используют для обнаружения всех остальных веществ из группы «летучих ядов». Двух-трех отгонов обычно бывает достаточно для качественного исследования.
При проведении исследования на группу «летучих» ядов, необходимо
обращать внимание на следующее:
1. Запах объекта (иногда это может дать какие-либо дополнительные ориентирующие данные). Правда, запах биологического объекта, как правило, маскирует запах летучего ядовитого вещества, но в некоторых случаях все же возможно определение запаха искомого соединения. Например, изоамиловый спирт придает объекту запах сивушных масел, нитробензол и синильная кислота запах горького миндаля.
2. Запах и внешний вид дистиллята. Перед выполнением химического исследования обязательно проводят наружный осмотр дистиллята, обращая внимание на его прозрачность или мутность, наличие капель на дне склянки или маслянистой пленки на поверхности жидкости, наличие характерного запаха.
Так изоамиловый спирт легче воды и плохо смешивается с ней, поэтому при содержании значительных количеств изоамилового спирта дистиллят обладает характерным раздражающим запахом сивушных масел и иногда содержит на поверхности маслянистые капли или даже два слоя этого вещества.
Присутствие в отгоне фенола можно обнаружить по характерному запаху карболовой кислоты и молочновидному помутнению, поскольку фенол плохо растворим в воде. При больших количествах фенола на дне приемника могут присутствовать бесцветные или розоватые капли (продукты окисления фенола).
Хлороформ и четыреххлористый углерод тяжелее воды и не смешиваются с ней, поэтому на дне приемника можно наблюдать бесцветные капли или слой этих веществ.
Исследование дистиллятов с целью идентификации веществ из группы «летучих ядов» традиционно строится на использовании микрохимических реакций. Так, реакция образования берлинской лазури, которая является высокочувствительной и специфичной для доказательства синильной кислоты имеет положительное судебно-химическое значение (т.е. на основании одной этой реакции можно дать положительное заключение о наличии в исследуемом объекте HCN). Она является особенно ценной еще и потому, что образующийся характерный осадок может быть представлен в качестве вещественного доказательства судебно-следственным органам.
Известно, что общей реакцией на алкилгалогениды является реакция отщепления органически связанного хлора. Реакция достаточно чувствительна, но не специфична. О таких реакциях принято говорить, что они имеют отрицательное судебно-химическое значение.
Заключение о присутствии того или иного из «летучих ядов» делается на основании комплекса реакций.
Кроме перегонки с водяным паром в токсикологической химии применять еще два метода дистилляции:
1. Микроперегонка
2. Микродиффузия
Микроперегонка. В последние годы исследование токсикологически важных «летучих» веществ все шире проводится методом газожидкостной хроматографии. Для изолирования в этом случае используется микроперегонка, поскольку количество объекта составляет 1-5 г.
Дата добавления: 2015-08-01; просмотров: 2541;