ИЗУЧЕНИЕ МЕТАБОЛИЗМА
Вторым подходом к решению вопроса о патогенезе осложнений диабета является изучение метаболизма в пораженных тканях (базальной мембране, периферических нервах). Такие исследования направлены на поиски метаболического звена, связывающего инсулиновую недостаточность и/или гипергликемию с осложнениями диабета [188]. Beisswenger и Spiro [189] сообщили о большем содержании оксилизина и оксипролина, а также глюкозы и галактозы в базальной мембране капилляров у больных диабетом (аутопсийный материал) по сравнению с таковым у лиц, не страдающих этим заболеванием. Поскольку повышенное содержание глюкозы в гликопротеинах может изменять их форму и ха-
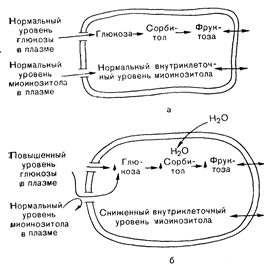
Рис.10—44. Схематическое изображение предполагаемого механизма, с помощью которого гипергликемия приводит к развитию диабетической нейропатии. а — шванновская клетка при нормальном уровне глюкозы и миоинозитола в плазме: б — то же при гипергликемии, которая обусловливает высокий уровень глюкозы в клетке, в силу чего накапливаются сорбитол и фруктоза. В ответ на осмотический эффект накопления сорбитола в клетку поступает вода. что приводит к ее набуханию. Показано также предполагаемое конкурентное ингибирование (гипергликемией) транспорта миоинозитола в шванновскую клетку, что .приводит к ее обеднению миоинозитолом (по Clements R. С. «Гг., Diabetes, 1979, 28, 604).
рактер упаковки, было выдвинуто предположение, согласно которому избыточное отложение глюкозы может также изменять проницаемость мембраны и играть роль в патогенезе протеинурии. Кроме того, известно, что отложение глюкозы в базальной мембране не зависит от инсулина, в силу чего можно было бы ожидать, что оно увеличивается по мере повышения уровня глюкозы в крови. Несмотря на привлекательность таких представлений, в ряде последующих работ не удалось подтвердить факт увеличения содержания глюкозы или оксилизина в базальной мембране почечных клубочков у больных диабетом [153]. Причина такого расхождения результатов неясна.
Изучали также химический состав и синтез миелина в ткани периферических нервов у животных с экспериментально вызванным диабетом. Gabby и соавт. [13] обнаружили повышенное содержание сорбитола в нервной ткани крыс с диабетом. Сорбитол представляет собой полигидроксилированный спирт (полиол), образующийся из глюкозы под действием фермента альдозоредуктазы (см. выше раздел «Углеводный обмен»). Этот фермент присутствует в нервной ткани и его активность не зависит от инсулина и не насыщается при повышенной концентрации глюкозы в крови. Осмотическая теория, предложенная Gabbay и соавт., заключается в том, что накопление сорбитола приводит к набуханию леммоцитов, в силу чего клетки погибают и демиелинизируются (рис. 10—44). Эту гипотезу подтверждают данные, свидетельствующие о снижении содержания сорбитола в нервной ткани под действием инсулинотерапии, а также о том, что ингибиторы альдозоредуктазы могут предупреждать накопление полиолов, как и замедление нервной проводимости у крыс, которым скармливают галактозу.
Однако предположение о набухании леммоцитов не нашло подтверждения при изучении содержания воды в цитоплазматических элементах нервной ткани [155].
В отличие от сорбитола, количество которого увеличивалось, содержание миоинозитола (полиоловое производное циклогексана) у крыс с экспериментальным диабетом снижалось (см. рис. 10— 44). Потенциальное значение этих данных определяется тем, что восстановление содержания миоинозитола путем его добавления к диете восстанавливало скорость проведения возбуждения по нервам у животных с тяжелым диабетом до нормы [190]. Однако при легкой форме диабета у животных содержание миоинозитола в нервах оставалось нормальным, а его добавка к корму не улучшала нервной проводимости. Кроме того, ограниченные данные в отношении человека не подтверждают ни уменьшения содержания миоинозитола в нервной ткани (материал аутопсии), ни закономерного клинического эффекта миоинозитола, получаемого с продуктами питания больными с нейропатией [155]. Интересно, что данные о повышении тканевого уровня миоинозитола привлекались для объяснения патогенеза и уремической нейропатии.
При диабете в нервах отмечались также изменения в содержании и синтезе миелина. Хотя химический состав миелина остается неизмененным, его общее количество у животных с экспериментальным диабетом снижено. Кроме того, уменьшается и включение лейцина в белки миелина, причем при добавлении инсулина in vitro оно увеличивается [191].
К патогенезу осложнений диабета может иметь отношение и тот факт, что у больных диабетом увеличивается количество гликозилированного гемоглобина (см. выше) [147]. Гликозилированные гемоглобины (гемоглобины A1а, A1b и A1c) представляют собой отрицательно заряженные минорные компоненты гемоглобина. Они отличаются от гемоглобина А присутствием глюкозы или глюкозофосфата, присоединенных к валину на аминоконце b-цeпи так, что появляется основание Шиффа, которое подвергается перегруппировке Амадори с образованием более стабильной кетоаминной связи (Рис. 10—45). Реакция конденсации глюкозы с остатком валина протекает без участия ферментов и зависит от окружающей концентрации глюкозы. Так, при недостаточно компенсированном. диабете общее содержание гемоглобина A1 превышает 12%, тогда как у здоровых лиц и у больных диабетом с менее выраженной гипергликемией его урвень обычно не достигает 6% [148, 149].. Особого упоминания заслуживают данные, свидетельствующие о том, что гликозилирование гемоглобина не ограничивается аминоконцом b-цепи, а захватывает и эпсилон-аминогруппу нескольких лизиновых остатков как a-, так и b-цепи [149]. Кроме того, лизиновые остатки в хрусталике при инкубации его с повышенными концентрациями глюкозы или глюкозо-6-фосфата также подвергаются, неферментативному гликозилированию [192а]. Таким образом, относительная неспецифичность мест гликозилирования позволяет предположить существование более общего феномена, который заключается в том, что гипергликемия обусловливает постсинте-
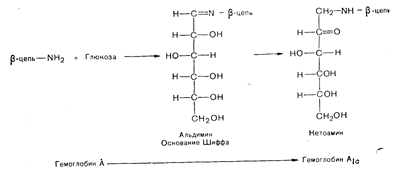
Рис. 10—45. Химические реакции, приводящие к неферментативному превращению гемоглобина А (НbА) в гликозилированный HbA1c. В первой реакции аминоконцевая (валин) аминогруппа b-цепи НbА конденсируется с глюкозой, образуя основание Шиффа. Затем оно подвергается перегруппировке Амадори с образованием более стабильного кетоамина HbA1с. тическую модификацию различных белков, включающих, возможно, белки базальной мембраны и миелина периферических нервов.
Против этих многочисленныхданных, свидетельствующих о химических изменениях в инсулинонезависимых тканях, говорит отсутствие прямых доказательств причинно-следственной связи между этими нарушениями и развитием микроангиопатии или нейропатии [182].
Еще одним метаболическим механизмом, с помощью которого дефицит инсулина мог бы обусловливать развитие осложнений диабета, является нарушение секреции гормона роста. У больных при недостаточной компенсации диабета в покое и даже при средней степени компенсации при физической нагрузке наблюдается гиперсекреция гормона роста. При нормализации гомеостаза глюкозы с помощью инсулина эти изменения исчезают [130]. Merimee [192b] обнаружил, что у карликов с дефицитом гормона роста и сосуществующим диабетом не развивается ни микроангиопатия,. ни нейропатия, несмотря на гипергликемию. Учитывая, однако, этиологическую гетерогенность диабета [110—112], неясно, сам ли по себе дефицит гормона роста или какие-либо другие генетические факторы оказывают у больных диабетом карликов защитное действие в отношении хронических осложнений этого заболевания.
Дата добавления: 2015-01-19; просмотров: 1198;