ПАТОФИЗИОЛОГИЯ СЕРДЕЧНО-СОСУДИСТОЙ СИСТЕМЫ 2 страница
способствующей повреждению клеток последнего иразвитию бляшки.
В 1974 г. появилась моноклональная теория,рассматривающая атероматозную бляшку как своеобразную «доброкачественную опухоль», образование которой вызвано вирусами и мутагенами. Автором этой теории явился американский ученый Е. Bendit, обративший внимание на то, что для атеросклеротических поражений характерна пролиферация гладкомышечных клеток. Близка по своей сути к предыдущей теории мембранная гипотезаD. Jackson и A. Gotto (1976), объясняющая причину пролиферации гладкомышечных клеток избыточным поступлением в них неэстерифицированного холестерина, который, встраиваясь в мембранные структуры клеток, способствует их гиперплазии.
В нашей стране под руководством академика А.Н.Климова активно развивается аутоиммунная теорияпатогенеза атеросклероза, согласно которой этот патологический процесс инициируют аутоиммунные комплексы, содержащие липопротеиды в качестве антигена. Аутоиммунные комплексы характеризуются следующими особенностями: 1) вызывают повреждение эндотелия и тем самым ускоряют проникновение липопротеидов крови в сосудистую етенку; 2) продлевают циркуляцию липопротеидов в крови; 3) задерживают окисление и экскрецию холестерина с желчью, т.е. способствуют развитию гиперлипопротеидемии; 4) проявляют цитоток-сическое действие, откладываясь и фиксируясь в стенке артерий.
Несмотря на существование различных теорий, наиболее популярной точкой зрения относительно патогенеза атеросклероза остается признание ключевой роли холестерина и атероген-ных липопротеидов.
Современные представления о роли холестерина и липопротеидов в атерогенезе.Как известно, в крови липиды содержатся в таких двух основных формах, как хиломикроны и липопротеиды.
ЛПНП и ЛПОНП осуществляют транспорт холестерина в клетки, участвуя в формировании атеросклеротических бляшек, поэтому их называют атерогенными.ЛПВП способны транспортировать холестерин из клеток эндотелия сосудов в печень, содействуя регрессии бляшек, в связи с чем их называют антиатерогенными.Эти различия в свойствах липопротеидов определяются их химическим составом. Так, в структуре
ЛПНП находится основное (около - 2/3) количество холестерина плазмы, в ЛПОНП - только 1/3 циркулирующего холестерина, а в ЛПВП -лишь следовые его количества. Кроме того, ате-рогенность липопротеидов во многом зависит от содержания в их структуре триглицеридов (они в основном содержатся в ЛПОНП), а также апоп-ротеинов и фосфолипидов (последних очень много в составе ЛПВП).
Транспорт холестерина из плазмы в сосудистую стенку осуществляется, главным образом, в виде ЛПНП, более половины которых попадает внутрь клеток эндотелия при помощи мембранных рецепторов, остальные - нерецепторным путем. Установлено, что указанные рецепторы взаимодействуют с апопротеинами, расположенными на поверхности ЛПНП. В лизосомах эндо-телиальных клеток ЛПНП распадаются, эфиры холестерина гидролизуются на холестерол и жирные кислоты, после чего жирные кислоты окисляются, а холестерол используется для «строительства» клеточных мембран. В ситуациях, когда ткани организма нуждаются в большом количестве холестерина (например, для синтеза клеточных мембран, стероидных гормонов, желчных кислот), активность клеточных ЛПНП-рецепторов возрастает, вследствие чего утилизация холестерина усиливается. В результате этого уменьшается содержание ЛПНП в крови и снижается вероятность транспорта холестерина в артериальную стенку. Если же ткани не нуждаются в дополнительных количествах холестерина, активность рецепторов ЛПНП снижается, концентрация ЛПНП в плазме возрастает, а вероятность формирования атеросклеротических бляшек увеличивается.
В отличие от липопротеидов низкой и оченг низкой плотности ЛПВП выполняют в организме антиатерогенную функцию. Согласно транспортной гипотезе В.Н.Титова, они осуществляют обратный транспорт холестерина из сосудов, органов и тканей, переводя его в другие липопротеиды или транспортируя прямо в печень, с последующим выведением с желчью в кишечник. В организме холестерин окисляется толькс в клетках печени. Следовательно, если содержание ЛПВП увеличивается, то одновременно усиливается окисление холестерина. Таким образом, чем больше в крови ЛПВП и холестерина в их составе, тем меньше вероятность развития атеросклероза и выше вероятность регресса атеросклеротических бляшек.
|
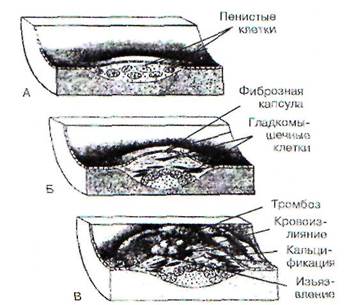
Рис. 132. Стадии атеросклероза: А - жировая полоска; Б - фиброзная бляшка; В - осложненное поражение
Вместе с тем сложную проблему атеросклероза нельзя свести к уровню холестерина и липо-протеидов в крови. Неверно строить рациональную терапию исключительно на диете по принципу малого содержания холестерина. Такой подход к патогенетически обоснованной профилактике упрощен, поскольку, по данным некоторых авторов [Шутова Н.Т., 1966], гиперхолес-теринемия отсутствует у 50% больных атеросклерозом.
Морфогенезатеросклероза. В формировании атеросклеротической бляшки - морфологической основы атеросклероза - важную роль играют как нарушения липидного обмена (дислипопротеи-демии), так и состояние сосудистой стенки. Бляшки могут расти вдоль сосуда, тогда они развиваются медленно, длительно и менее опасны, но могут располагаться и поперек сосуда -такие бляшки часто называют «летальными», поскольку даже единичные образования подобного типа могут привести к сосудистой катастрофе.
Предшественниками бляшки часто являются зоны липидной инфильтрации интимы, так называемые липидные полоски,через которые в сосудистую стенку проникают моноциты. В сосудистой стенке моноциты трансформируются в макрофаги, имеющие рецепторы к ЛПНП. В
процессе переполнения этих клеток фагоцитированными липопротеидами они превращаются в пенистые клетки. Имеются данные, что пенистыми клетками могут становиться и переполненные липидами гладкомышечные клетки. Скопления пенистых клеток и составляют основу липидных полосок. Пенистые клетки могут разрушаться, высвобождая биологически активные вещества, стимулирующие пролиферацию ГМК и привлекающие их в субэндотелиальный слой из глубжележащих участков сосудистой стенки. В результате скопления ГМКнаблюдается образование небольших выпячиваний эндотелия в просвет сосуда.
В процессе дальнейшего развития бляшек в них появляются соединительнотканные элементы: коллагеновые и эластические волокна, приводящие к уплотнению - склерозу. Этот процесс поддерживается за счет выделения из макрофагов медиаторов клеточного иммунитета и ростовых факторов, стимулирующих аутоиммунные реакции в интиме и пролиферацию фиброблас-тов. В результате образуется плотная фиброзная бляшка.
Конечным этапом формирования бляшек является образование их «осложненных» форм (рис. 132). Выступающая в просвет сосуда ате-росклеротическая бляшка насыщается солями кальция и нарушает ламинарный поток крови, который в этом месте становится турбулентным. Такая бляшка пропитывается липидами и становится рыхлой. Очевидно, что плотная фиброзная бляшка является потенциально менее опасной, чем ее рыхлая «осложненная» форма, которая вследствие слущивания покрывающего ее эндотелия, кальцификации и происходящего в ней клеточного распада таит угрозу образования пристеночного тромба или разрыва сосуда с кровоизлиянием.
14.2.2. Артериальные гипертензии
Артериальная гипертензия (АГ) - повышение внутрисосудистого давления в артериях большого круга кровообращения.Возникает в результате усиления работы сердца, увеличения периферического сопротивления или сочетания этих факторов. По данным комитета экспертов ВОЗ, артериальная гипертензия встречается у 15-25% взрослого населения, частота ее увеличивается с возрастом и регистрируется более чем у 50% людей старше 65 лет.
 Артериальная гипертензия длительное время протекает без явных клинических симптомов. Однако достаточно скоро АГ может привести к возникновению острых нарушений мозгового кровообращения (транзиторная ишемическая атака, ишемический или геморрагический инсульт) и развитию гипертрофии миокарда. Кроме того, АГ является фактором риска атеросклероза и возникновения инфаркта миокарда.
Артериальная гипертензия длительное время протекает без явных клинических симптомов. Однако достаточно скоро АГ может привести к возникновению острых нарушений мозгового кровообращения (транзиторная ишемическая атака, ишемический или геморрагический инсульт) и развитию гипертрофии миокарда. Кроме того, АГ является фактором риска атеросклероза и возникновения инфаркта миокарда.
По своему происхождению артериальная гипертензия бывает первичной и вторичной.
Первичная артериальная гипертензия (гипертоническая болезнь) - это стойкое повышение АД, не связанное с органическим поражением органов и систем, регулирующих сосудистый тонус. Распространенным названием первичной гипертензии является еще термин «эс-сенциальная гипертония», что означает неясность ее этиологии. На долю гипертонической болезни приходится 90-95% от общего числа артериальных гипертензии.
Вторичная артериальная гипертензия - это повышение АД, представляющее собой лишь симптом другого диагностически подтвержденного заболевания (гломерулонефрит, стеноз почечных артерий, опухоль гипофиза или надпочечников и т.д.). В связи с этим вторичная гипертензия называется еще симптоматической. На долю подобного рода нарушений сосудистого тонуса приходится в среднем 5-10%. Виды симптоматических гипертоний: эндокринная, нефро-генная, гемодинамическая, нейрогенная и лекарственная.
Симптоматические артериальные гипертензии
Нефрогенные артериальные гипертензии.
Возникают при врожденных или приобретенных заболеваниях почек (аномалии развития почек, гломерулонефрит, пиелонефрит и др.), сопровождающихся расстройствами регионарного кровообращения (вазоренальная или реноваскулярная артериальная гипертензия) и поражением почечной паренхимы (ренопаренхиматозная или ре-нопривная артериальная гипертензия). Нарушения внутрипочечного кровотока вызывают ишемию почек, которая выступает в роли «пускового механизма», активирующего секрецию ренина в юкстагломерулярном аппарате (ЮГА). Следует отметить, что нарушение функции этой си-
стемы в большей степени характерно для вазо-ренальной (симптоматической) гипертензии, хотя и при эссенциальной гипертонии ренин-ангио-тензин-альдостероновая система (РААС) активно вовлекается в патогенетическую цепь стойкого повышения артериального тонуса.
Ренин поступает в кровь и вызывает энзима-тическое расщепление плазменного белка анги-отензиногена, относящегося к а2-глобулинам. В результате этого образуется декапептид ангио-тензин-I, который под влиянием ангиотензин-превращающего фермента (ангиотензинконвер-таза) переходит в октаиептид ангиотензин-П, представляющий собой один из самых сильных вазоконстрикторов. Надо особо отметить, что ангиотензин-П вызывает стойкое и длительное повышение АД, что связано с его достаточно медленным ферментативным расщеплением.
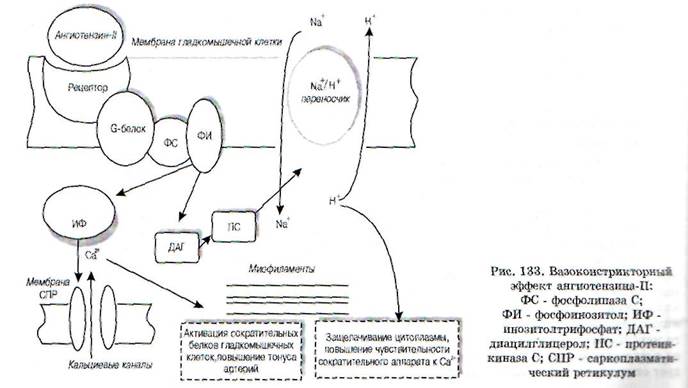
В настоящее время молекулярный механизм действия ангиотензина-П на сосудистую стенку хорошо изучен. Установлено, что он взаимодействует со специфическими рецепторами, расположенными на сарколемме гладкомышечных клеток. Активация этих рецепторов вызывает цепочку событий, очень сходную с теми, которые наблюдаются в условиях активации а-адре-норецепторов. Как показано на рис. 133, после взаимодействия ангиотензина-Ц с рецептором наблюдается опосредованная через G-белки активация фосфолипазы С, что ведет к усилению гидролиза фосфоинозитидов с образованием внутриклеточных мессенджеров: инозитолтрифосфата и диацилглицерола. Инозитолтрифосфат мобилизует Са2+ из депо в саркоплазматический ре-тикулум и вызывает сокращение гладкомышечных клеток. ДАГ активирует протеинкиназу С, которая фосфорилирует белки протон-натриевого (Na+/H+) переносчика, расположенного на клеточной мембране. В результате наблюдается усиленное поступление Na' в цитоплазму и, наоборот, выведение протонов во внеклеточное пространство, приводящее к защелачиванию саркоплазмы (увеличение рН). В условиях защелачи-вания цитоплазмы заметно повышается чувствительность сократительного аппарата гладкомышечных клеток к ионам кальция и, как следствие, усиливается их способность к сокращению даже при низком уровне внутриклеточного кальция [Са2*^. Именно такая последовательность событий обеспечивает вазоконстрикторный эффект ангиотензина-П.
Однако ангиотензин-И не только повышает тонус артерий, но и оказывает митогенное действие, вызывая усиленную пролиферацию глад-комышечных клеток и утолщение сосудистой стенки. По этой причине ангиотензин-И называют еще ростовымфактором. Указанный эффект опосредуется через активацию протеинки-назы С, тирозинкиназы и вызываемое ими фос-форилирование регуляторных белков. Функциональные и морфологические изменения артерий, индуцированные ангиотензином-П и другими эндогенными биологически активными веществами, получили название ремоделирова-ниясосудистой стенки.
В последние годы рецепторы ангиотензина-И были обнаружены и в надпочечниках. Стимуляция этих рецепторов вызывает усиление секреции альдостерона, который индуцирует задержку Na"1 и воды в организме. Такие изменения водного и солевого обмена ведут к увеличению объема циркулирующей крови и повышению АД.
Таким образом, ангиотензиновое звено патогенеза артериальной гипертонии включает в себя три основных компонента: 1) повышение тонуса артерий; 2) ремоделирование сосудистой стенки; 3) усиление секреции альдостерона.
Следует отметить, что одностороннее наруше-
ние почечного кровообращения,как правило, приводит лишь к преходящей гипертензии. Однако если при этом у экспериментального животного удалить вторую («нормальную») почку, развивается стойкое повышение АД. Дело в том, что паренхиматозная ткань почек секретирует не только ренин, но и ряд веществ, которые препятствуют повышению АД, в частности простаг-ландины, способные оказывать прямое сосудорасширяющее действие, ангиотензиназу, расщепляющую ангиотензин-И, и др. В результате первоначальное повышение АД, вызванное гипоксией одной почки и выбросом ренина, сменяется его нормализацией. Именно этим можно объяснить тот факт, что удаление обеих почек в эксперименте вызывает возникновение стойкой гипертензии, получившей название реноприв-ной.
Симптоматические артериальные гипертензии эндокринного происхождения наблюдаются главным образом при следующих заболеваниях: фе-охромоцитоме, первичном альдостеронизме (синдром Конна), болезни и синдроме Иценко - Ку-шинга, тиреотоксикозе.
Феохромоцитома.Так называется опухоль мозгового вещества надпочечников, продуцирующая значительные количества катехоламинов
|
Часть III. ПАТОФИЗИОЛОГИЯ ОРГАНОВ И СИСТЕМ
(адреналин, норадреналин, дофамин), концентрация которых у больных с феохромоцитомой в крови и моче увеличивается в 10-100раз. Именно избыточной секрецией катехоламинов и определяется гипертензионный синдром при данном заболевании. При феохромоцитоме выделяют три варианта артериальной гипертензии: стабильную, пароксизмальную (кризовую) и смешанный тип с пароксизмами на фоне стабильного повышения АД.
Первичный альдостеронизм (синдром Кон-на).Морфологическим субстратом данного заболевания чаще всего являются солитарные или (реже) множественные аденомы и идиопатичес-кая гиперплазия клубочковой зоны коры надпочечников, секретирующие альдостерон. В настоящее время идентифицированы уже три гена, регулирующих секрецию альдостерона. Мутация любого из них ведет к гиперальдостеронизму (так называемый семейный, или первичный, гипе-ральдостеронизм) и АГ. Основные симптомы болезни определяются гиперпродукцией альдостерона - минералокортикоиды способствуют ре-абсорбции в почках гидратированных ионов натрия, что ведет к задержке в организме воды и увеличению объема циркулирующей крови, а следовательно, к подъему АД и формированию АГ.
Болезнь и синдром Иценко - Кушинга.Приводят к повышению в крови уровня глюкокор-гикоидов, которые при данной патологии играют решающую роль в формировании АГ. Первичное или вторичное (под влиянием АКТГ) повышение секреции глюкокортикоидов вызывает увеличение плотности адренорецепторов, локализованных в сердце и сосудах, а также повышение их чувствительности к катехоламинам; стимуляцию продукции ангиотензиногена в печени. Вслед за повышением адренореактивнос-ти сердца и сосудов наблюдается увеличение тонуса сосудов и увеличение сердечного выброса. Результатом этих гемодинамических эффектов является увеличение АД.
Гипертиреоз.Возникает при гиперфункции щитовидной железы, в результате чего повышается уровень тироксина и трийодтиронина в крови. При этом подъем АД происходит вследствие сочетанного возрастания периферического сопротивления артерий и увеличения минутного объема сердца. Последний эффект обусловлен тирок-синзависимой тахикардией.
Гемодинамические гипертензии.Возникают
в результате изменений в сердце или крупных сосудах. Чаще развивается систолическая гипертензии с увеличением пульсового давления. В одних случаях (коарктация аорты, неспецифический аортоартериит) гипертензии является региональной, в других - носит системный характер.
Коарктация аорты- врожденное сегментарное сужение нисходящей части грудной аорты, создающее два режима кровообращения в большом круге: гипертензию верхней половины туловища и гипотензию нижней. У мужчин встречается в 4 раза чаще, чем у женщин. Неспецифический аортоартериит представляет собой системное сосудистое заболевание аутоиммунного генеза, приводящее к повышению ригидности аорты и магистральных артерий, а также к их стенозированию. Одним из клинических проявлений этого заболевания служит артериальная гипертензии, вызванная повышением периферического сосудистого сопротивления. Это патогенетическое звено играет важную роль также при потере эластичности сосудистой стенки у пациентов с распространенным атеросклерозом аорты и магистральных артерий.
Нейрогенные гипертензии.Развиваются при опухолях, ушибах и сотрясениях головного мозга, менингитах, менингоэнцефалитах, ишемии головного мозга, вызванной атеросклерозом ветвей брахиоцефальных артерий или сдавлением позвоночных артерий, которые связаны с остеохондрозом шейно-грудного отдела позвоночника. Патогенез подобных гипертоний обусловлен изменением тонуса высших вегетативных центров, регулирующих уровень АД.
Лекарственные гипертензии.Многие лекарственные препараты, воздействуя на разные звенья регуляции АД, могут вызывать его повышение. Следует прежде всего обратить внимание на глюкокортикоидные гормоны, широко применяемые в терапии различных системных заболеваний.
Гипертоническая болезнь
Этиология гипертонической болезни.Первостепенное значение в возникновении гипертензии имеет длительное психоэмоциональное перенапряжение.Об этом свидетельствуют частые случаи развития первичной гипертензии у лиц, переживших ленинградскую блокаду, а также у людей «стрессовых» профессий. Особую роль
играют отрицательные эмоции. В отличие от представителей животного мира современный цивилизованный человек часто не имеет возможности «погасить» свое эмоциональное возбуждение двигательной активностью. Это способствует длительному сохранению в коре головного мозга очага застойного возбуждения и развитию артериальной гипертензии. На этом основании Г.Ф.Ланг и А.Л.Мясников назвали гипертоническую болезнь болезнью неотреагированных эмоций.
Гипертоническая болезнь - это «болезнь осени жизни человека, которая лишает его возможности дожить до зимы». Так писал академик А.А. Богомолец, подчеркивая тем самым предрасполагающую роль возрастав ее происхождении. Однако нередко первичная гипертензия развивается и в молодом возрасте (ювенильная форма гипертензии). Важно при этом отметить, что до 40 лет мужчины болеют чаще, чем женщины, а после 40 лет соотношение приобретает противоположный характер.
Важная роль в этиологии первичной гипертензии отводится наследственности. На роль генетических факторов указывают и факты высокой конкордантности по гипертонической болезни у однояйцевых близнецов, а также существование линий крыс, предрасположенных к развитию гипертензии (SHR - spontaneously hypertensive rats). Из 9 уже известных канди-датных генов АГ в последнее время особое внимание привлекает ген эндотелиальной NO-син-тетазы. NO-синтетаза, локализованная в стенке артерий, катализирует синтез эндотелийрелак-сирующего фактора, который обеспечивает ва-зодилатацию и снижение АД. По вполне понятным причинам аномалия гена, кодирующего синтез этого фермента, может обусловить развитие АГ.
Успешное экспериментальное моделирование «солевой гипертензии» говорит в пользу этиологической роли избыточного потребления соли.Однако правильнее, видимо, считать этот фактор не столько главной причиной, сколько фактором риска. Считают даже, что длительное потребление более 5 г соли в день способствует развитию гипертензии только у лиц, имеющих наследственное предрасположение к ней.
Патогенез гипертонической болезни.Несмотря на то, что эссенциальная и вторичная АГ существенно различаются по этиологическим фак-
торам, механизмы артериальной гипертензии, независимо от вызывающих ее причин, имеют много общего. Согласно концепции, разработанной Г.Ф.Лангом и А.Л.Мясниковым, «нервное перенапряжение при гипертонической болезни реализуется в расстройстве трофики определенных мозговых структур, управляющих артериальным давлением», прежде всего тех областей коры больших полушарий и подкорковых центров (гиппокамп, миндалевидное тело), экспериментальное раздражение которых вызывает повышение артериального давления. Так, было установлено, что ишемия головного мозга, вызванная у кролика перевязкой сонных артерий, способствует возникновению АГ. У высокоорганизованных животных (собаки, обезьяны) удалось получить рефлексогенную гипертензию путем «сшибки» пищевого и оборонительного рефлексов. В этом случае гипертензия явилась следствием невроза. Непосредственный механизм повышения артериального давления связан с возникновением очага застойного возбуждения (патологической доминанты, по терминологии А.А. Ухтомского) вегетативных центров головного мозга, в первую очередь сосудодвигатель-ного центра. Вазомоторные импульсы, возникающие в ядрах гипоталамуса, поступают в ядра продолговатого мозга, откуда по симпатическим путям передаются на сосуды резистивного типа, вызывая повышение их тонуса.
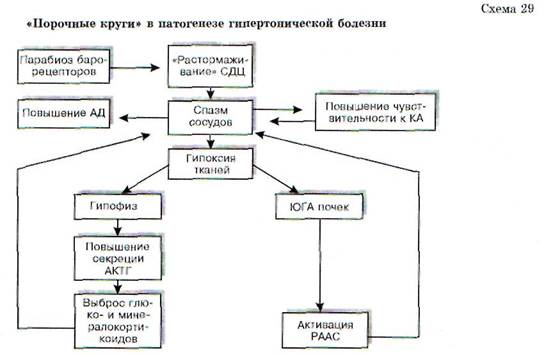
Важную роль в патогенезе гипертонической болезни играет «растормаживание» сосудодви-гательного центра (СДЦ), расположенного в продолговатом мозге. В норме его активность реф-лекторно подавляется импульсами, поступающими от синокаротидной зоны и рецепторов дуги аорты. При гипертонической болезни активность этого центра очень часто повышена.
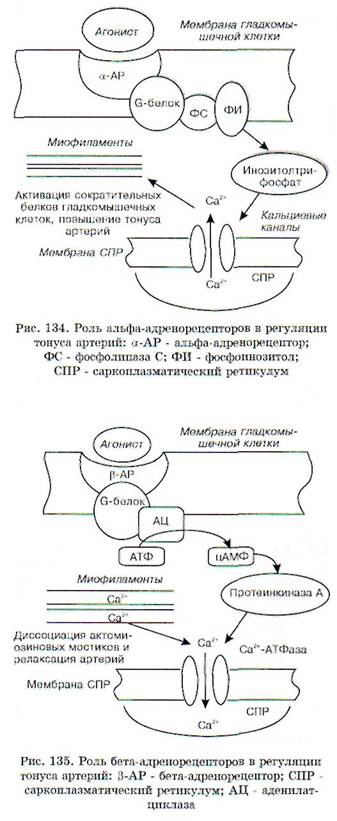
Другим слагаемым гипертензивного эффекта, наблюдаемого при возбуждении высших вегетативных центров головного мозга, является выброс катехоламинов (адреналина и норадренали-на) из мозгового вещества надпочечников в кровоток. Развитие гипертензии при этом наблюдается тогда, когда а-адренергические эффекты этих гормонов суммарно превышают присущее им влияние на Р-адренергические рецепторы (рис. 134, 135).
Рассмотренные процессы формируются на первой стадии гипертонической болезни, которая называется транзиторной( преходящей) и
|
клинически характеризуется непродолжительными эпизодами повышения АД.
В дальнейшем снижается лабильность ипоявляется инертность нервных структур, составляющих сосудодвигательный центр, и развивается вторая стадия болезни - стабильная. В этой стадии высокий уровень АД поддерживается длительно. Стимулирующим влиянием на сосудодвигательный центр во второй стадии гипертонической болезни обладают не только специфические раздражители, но и«посторонние», исходящие из соседних структур импульсы, даже подпорогового уровня. В происхождении стабильного повышения тонуса сосудов имеют значение формирующиеся в этой стадии «порочные круги». В результате повышения АД развивается парабиоз барорецепторов сосудов и выпадает их тормозный контроль над нейронами сосудодви-гательного центра (схема 29). В итоге тонус сосудов повышается еще сильнее. Спазм сосудов приводит к гипоксии юкстагломерулярного аппарата почек и активации ренин-ангиотензин-альдостероновой системы. В свою очередь, ише-мическая стимуляция аденогипофиза реализуется в секреции АКТГ и, следовательно, повышении содержания в крови гормонов коры над-почеников (минерало- и кортикостероидов). Поэтому высокий тонус сосудов поддерживается продолжительное время. В механизме гипертонии также играет роль повышение чувствительности стенки сосудов к катехоламинам (КА), идаже небольшие дозы адреналина оказывают выраженный вазоконстрикторный эффект.
Таким образом, определяющую роль в патогенезе АГ играют изменения нейрогуморальной регуляции сосудистого тонуса. Конечным звеном этого патологического процесса является изменение функциональной активности ионотранс-портирующих систем плазматической мембраны, что ведет к перегрузке клеток ионами кальция и патологическому повышению тонуса кровеносных сосудов. Такая концепция патогенеза артериальной гипертензии была выдвинута Ю.В.Постновым и С.Н.Орловым, которые назвали ее мембранной концепцией.
Третья стадия гипертонической болезни - стадия органных изменений.В начале этой стадии можно обнаружить гипертрофию левого желудочка. В дальнейшем развивается кардиосклероз и присоединяется сердечная недостаточность. Со стороны внутренних органов отмечаются ишемические повреждения, которые индуциро-
Глава 14 / ПАТОФИЗИОЛОГИЯ СЕРДЕЧНО-СОСУДИСТОЙ СИСТЕМЫ
|
 ваны морфологическими изменениями стенки ятельными по причине быстрого развития ги-
ваны морфологическими изменениями стенки ятельными по причине быстрого развития ги-
сосудов (гиалиноз, склероз, атеросклероз). Наи- пертензии. Криз, как правило, проявляется силь-
более характерным является повреждение парен- нейшими головными болями, тошнотой, рвотой
химы почек с развитием хронической почечной и может закончиться гибелью пациента от ин-
недостаточности, задержкой жидкости в организ- фаркта миокарда или мозгового инсульта, по
ме и прогрессированием АГ. этому требует экстренного медикаментозного
Артериальная гипертензия независимо от вмешательства, направленного на нормализацию
причин, ее обусловивших, протекает длительно АД.
и имеет тенденцию к неуклонному прогрессиро-
ванию. Однако относительно «спокойное» раз- 14.2.3. Легочная гипертензия
витие заболевания может осложняться возник- тт /тт\
, Легочная гипертензия (ЛГ), или артериаль-
новением так называемых гипертензивных (ги- _._ ___ __ _____ „„«„
r v ная гипертензия малого круга кровообраще-
пертонических) кризов.
„ - *■ - ния, - это патологическое состояние, характе-
Гипертензивныи криз представляет собой
г д п ризующееся повышением систолического, ди-
внезапное повышение АД, сопровождаемое по-
_ „ астолического и среднего давления в легочной
явлением или усугублением уже имеющейся „ ,
„ / • „ артерии. В малом круге кровообращения, как и
церебральной и/или кардиальнои симптомати- \» . тт
„ . тт в в большом, АД может повышаться при усиле-
ки. Систолическое АД при этом очень быстро , , -
Тпг» ото _____ нии работы сердца (увеличение минутного объе-
может повыситься до 190-270 мм рт.ст., а диас- \ /
.„„..л „ тт ~ ма правого желудочка) и/ или росте сопротивле-
толическое - до 120-160 мм рт.ст. Прямой зави- v A
. тт ния легочных сосудов. Однако артерии малого
симости между уровнем АД и выраженностью - Г ^
й m круга кровообращения обладают большой рас-
симптоматики криза не наблюдается. Тяжесть уу у у ^ м у. п
__ -___ тяжимостью, и для заметного повышения АД
криза определяется не столько величиной подъе- _ -
„АТт„л_______________ ,„__________ ?,„ „„„л„„ необходимо увеличение минутного объема кро-
ма АД, сколько степенью развившейся дисфун- - п
кции жизненно важных органов - головного моз- вообращения как минимум в 3 раза или умень-
га и сердца. Гипертензивный криз относится к шение емкости сосудов малого круга кровообра-
разряду ургентных (неотложных) ситуаций, по- ще»ия пРимерно на 2/3.
Дата добавления: 2015-03-19; просмотров: 1114;