Раздел 2. ОСНОВЫ ТЕОРИИ ФОРМИРОВАНИЯ КРИСТАЛЛИЧЕСКОЙ СТРУКТУРЫ ОТЛИВОК 2 страница
При атомно-гладкой поверхности зародыша (рис. 2.10, а) возможен только послойный рост кристалла путем образования на гранях двумерных зародышей, которые разрастаются
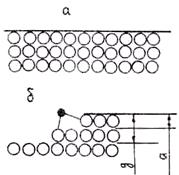

Рис. 2.10. Типы морфологии поверхности растущего Рис. 2.11. Рост кристалла путем присоединения
кристалла: а — атомно-гладкая поверхность; отдельных атомов (а) и двумерных зародышей (б)
б — ограненная поверхность
затем вдоль этих граней. Рост путем присоединения отдельных атомов в данном случае энергетически неустойчив, так как каждый адсорбированный поверхностью атом имеет только одного ближайшего «соседа» и, следовательно, только одну связь (рис. 2.11, а). В таких условиях он не может устойчиво закрепиться на поверхности. Поэтому рост может происходить только путем образования и закрепления тонких, одно-, двух- и трехатомных по толщине, плоских образований атомов, называемых двумерными зародышами.
На рис. 2.12 приведена схема двумерного зародыша на грани растущего кристалла. При образовании такого зародыша поверхность кристалла увеличивается на ΔS = 4×2x×d = 8x×d, т. е. на величину поверхности четырех боковых граней. Соответствующее возрастание поверхностной энергии равно As = 8xds = 8×xs', где s' = `ds — поверхностная энергия, приходящаяся на единицу периметра зародыша толщиной d. Если энергию Гиббса зародыша с основанием 1 см2 обозначить j2, а энергию Гиббса, приходящуюся на то же число атомов жидкости, — j1, то для полного изменения энергии при зародышеобразовании можно написать следующее выражение:
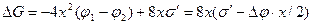 .
.
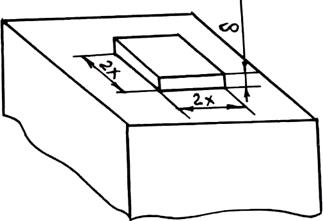
Рис. 2.12. Схема образования двумерного зародыша на грани кристалла
Подставив в это выражение Δj = j1 — j2 = ΔH`×ΔT/Ткр (см. гл. 2.1), где ΔH' — теплота плавления зародыша с основанием 1 см2, получаем
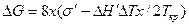 .
.
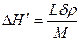 ;
; 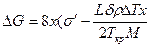 .
.
Исследуем эту зависимость на экстремум:
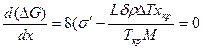 .
.
Из этого уравнения находим выражение для вычисления критического размера зародыша хкр, при котором ΔG достигает максимума:
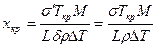 .
.
Работа образования этого зародыша вычисляется по формуле
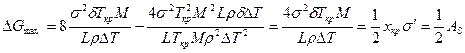
Сравнение полученных формул для вычисления размера и работы образования двумерного критического зародыша с соответствующими выражениями для трехмерного зародыша (см. гл. 2.1) показывает, что критический размер двумерного зародыша в 2 раза меньше, чем трехмерного, меньше и работа образования критического двумерного зародыша. Вероятность образования в единице объема двумерного зародыша равна
 .
.
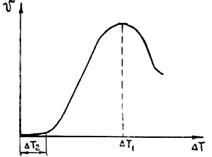
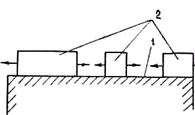
Рис. 2.13. Зависимость линейной скорости Рис. 2.14. Схема одновременного роста нескольких
роста кристалла от переохлаждения двумерных зародышей:
/—грань кристалла; 2—зародыши
Вероятность присоединения к зародышу в единицу времени хотя бы одного атома равна р2 = с2×ехр(—U/kT). Вероятность образования центра кристаллизации вычисляется по формуле
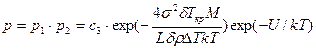 ,
,
где с3 = с1×с2 — некоторая постоянная для данного вещества величина.
Скорость роста кристалла пропорциональна вероятности р, т. е.
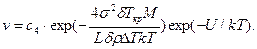
Характер зависимости линейной скорости роста от переохлаждения ΔТ показан на рис. 2.13.
Величина интервала метастабильности при росте кристаллов ΔТ2 значительно меньше, чем ΔТ1 при зарождении. При этом, как правило, реализуется только восходящая ветвь кривой (см. рис. 2.13). Зависимость можно аппроксимировать следующей функцией:
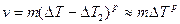 ,
,
где показатель степени 1 £ р £ 2. Двумерный зародыш быстро растет, последовательно присоединяя к своим боковым граням ряды атомов так, что слой, образованный зародышем, покрывает грань кристалла. После заполнения грани наступает пауза, длительность которой t ~ ехр(4s`хкp/kT) обратно пропорциональна вероятности р1. Затем наращивается новый слой и т. д. У кубического кристалла в идеальном случае все шесть граней перемещаются с одинаковой скоростью в направлениях своих нормалей.
Когда грань кристалла достаточно велика, длительность ее заполнения t значительно больше времени ожидания появления нового зародыша t. В этом случае возможны образование и независимый рост двух или нескольких зародышей (рис. 2.14).
2. На атомно-шероховатых поверхностях кристаллов могут надежно адсорбироваться отдельные атомы жидкости (рис. 2.10, б). При этом происходит так называемый нормальный рост кристаллов, когда атомы расплава беспорядочно присоединяются к любым точкам поверхности. В этом случае грань кристалла перемещается однородно по нормали к самой себе. Соответствующий расчет показывает, что линейная скорость роста в данном случае пропорциональна переохлаждению:
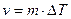 ,
,
где m = n×L×M×exp(—U/kT)/(T2nep×R); R — универсальная газовая постоянная; n — частота колебаний атомов в жидкости.
3. Механизм роста существенно изменяется, если на поверхности растущего кристалла имеются какие-нибудь выступы, которые облегчают закрепление частиц жидкости на поверхности кристалла. Очень часто такие выступы образуются в местах выхода винтовых дислокаций. Кристаллы, на гранях которых имеются ступеньки, возникшие в результате выхода винтовых дислокаций, растут послойно путем присоединения атомов расплава к этим ступенькам. Не приводя вывода, запишем формулу для вычисления линейной скорости в этом случае:
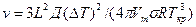 ,
,
где s — межфазное натяжение на границе кристалл — жидкость; Vm — молярный объем жидкости; Д — коэффициент диффузии.
Из формулы видно, что линейная скорость роста v пропорциональна (ΔT)2. Дислокационный послойный рост происходит при значительно меньших переохлаждениях, чем рост двумерных зародышей. Интервал метастабильности по росту в этом случае равен нулю.
На рис. 2.15 приведены результаты исследования механизма роста в зависимости от величины переохлаждения на фронте кристаллизации. Видно, что при малых переохлаждениях рост осуществляется путем образования двумерных зародышей, при больших переохлаждениях
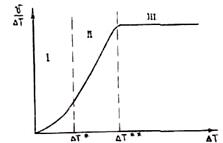
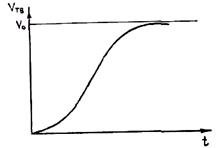
Рис. 2.15. Зависимость механизма роста кристаллов от Рис. 2.16. Кинетическая кривая криcталлизации
величины переохлаждения на фронте кристаллизации:
I — зона образования двумерных зародышей;
II — промежуточная зона;
III — зона нормального роста кристаллов
— по нормальному механизму. При этом фронт не огранен, т. е. имеет вид атомно-шероховатой поверхности. В промежуточной зоне переохлаждения рост может осуществляться по дислокационному механизму. В реальных условиях литья, как правило, реализуется нормальный механизм роста.
ГЛАВА 2.4. КИНЕТИКА КРИСТАЛЛИЗАЦИИ. ФОРМУЛА КОЛМОГОРОВА
Рассмотренные выше характеристики процесса кристаллизации—скорость возникновения центров кристаллизации п и линейная скорость роста кристаллов v — являются важнейшими кристаллизационными характеристиками, определяющими кинетику кристаллизации. Математической величиной, описывающей кинетику кристаллизации, является временная зависимость объема закристаллизовавшейся жидкости.
Решение задачи о кинетике нарастания твердой фазы при кристаллизации было впервые выполнено А. Н. Колмогоровым. Используя аппарат теории вероятности, можно получить следующую формулу:
 ,
,
где n(t) = v(т) — скорость возникновения центров кристаллизации и линейная скорость их роста как функции времени; V0 — начальный объем расплава. Формула справедлива для сферического зародыша. Если принять в ней n = const и v = const, то после интегрирования нетрудно получить формулу Колмогорова
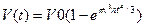 . (2.3)
. (2.3)
Графически она описывается кривой, показанной на рис. 2.16. Видно, что на начальных и конечных временных участках наблюдается незначительный темп нарастания твердой фазы. Замедление кристаллизации в конце объясняется тем, что растущие кристаллы начинают соприкасаться друг с другом при малом и непрерывно уменьшающемся количестве жидкости. Так как в конце кристаллизации объем жидкости мал, то основное положение вывода V0 >> Vкр не соблюдается. Поэтому из формулы вытекает неправдоподобный результат, что продолжительность полного затвердевания объема равна бесконечности. При этом погрешность формулы тем больше, чем меньше кристаллов и чем больше размер зерна. Принято считать, что формула обеспечивает расчет с погрешностью не более 1 %, если выполняется условие V0(n/v)3/4 > 200. Анализ показывает, что это условие, как правило, соблюдается при затвердевании реальных сплавов.
В обобщенном виде уравнение кинетики кристаллизации можно записать следующим образом:
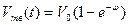 ,
,
где 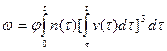 ; (j — коэффициент, учитывающий конфигурацию кристаллита (j = 4p/3 для шара и j = 8 для кубических кристаллитов).
; (j — коэффициент, учитывающий конфигурацию кристаллита (j = 4p/3 для шара и j = 8 для кубических кристаллитов).
Рассмотрим кинетику кристаллизации при объемном затвердевании. Возьмем некоторый объем сплава Vo, который охлаждается при малой интенсивности теплообмена с внешней средой. В этих условиях можно пренебречь перепадом температур по объему и рассматривать, что кристаллизация происходит путем равномерного выделения по всему объему твердой фазы. Такое затвердевание называют чисто объемным. Пусть при t = 0 температура расплава равна T = Tкр—ΔT1, где ΔT1 — интервал метастабильности по зарождению кристаллов. Напишем уравнение теплового баланса:
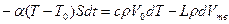 ,
,
где a — коэффициент теплоотдачи с наружной поверхности объема; S — поверхность охлаждения; T0—температура среды; L, с и р — удельная теплота кристаллизации, теплоемкость и плотность расплава; Vтв — объем металла, затвердевшего за время t; dT—изменение температуры за время dt. Для оценки зависимости параметров п и v от переохлаждения примем v = mΔT = m(Tкр – T), n = n0(ΔT – ΔT1)2 = n0(Tкp – T – ΔTl)2 приΔT>ΔT1 и n = 0, если ΔT £ ΔT1. Проинтегрируем это уравнение при начальном условии: при t = 0 Vтв = 0 и T = Tкр – ΔT1. Кроме того, в левой части уравнения примем T = Ткр ввиду малой величины переохлаждения ΔT при кристаллизации металлов.
 ,
,
Подставив в это уравнение выражение для Vтв (см. формулу Колмогорова), получаем интегральное уравнение, описывающее изменение температуры металла при его объемной кристаллизации:
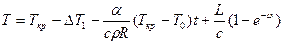 . (2.4)
. (2.4) 
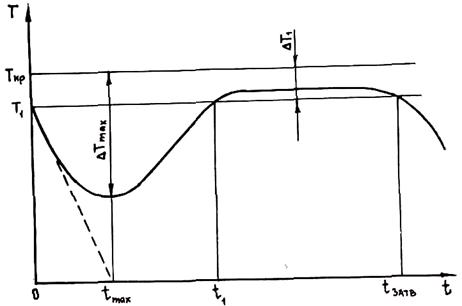
Рис. 2.17. Изменение температуры расплава при его объемной кристаллизации
Решить это уравнение возможно только с применением численных методов на ЭВМ. Изменение температуры во времени показано на рис. 2.17 (расчетные данные). Подобный характер температурной кривой подтверждается многочисленными экспериментами.
Если бы в расплаве не выделялась теплота кристаллизации, то температура уменьшалась бы в соответствии со штриховой линией на рис. 2.17, которая отвечает уравнению T = Tкр – ΔT1 - (Ткр – Т0)×t×a /(c×r×R). Однако выделяющаяся теплота кристаллизации замедляет темп охлаждения, и в момент t = tmax dT/dt = 0, что соответствует минимуму температуры, или максимуму переохлаждения (ΔT = Tкр – Т). При t > tmax dT/dt > 0, а температура сплава повышается, пока переохлаждение не станет меньше интервала метастабильности по зарождению ΔT1. При t > t1 ΔT < ΔТ1 и зарождение новых кристаллов в расплаве прекращается. Далее будет происходить только рост образовавшихся в период 0 < t < t1 центров до полного затвердевания металла при t = tзатв. Этот подъем температуры называется рекалесценцией.
Приближенное решение уравнения, выполненное с использованием метода итераций, позволило получить следующие формулы для расчета параметров температурной кривой:
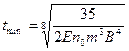 ;
;
 ;
;
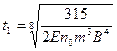 ;
; 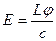 ;
;  .
.
Для вычисления количества центров кристаллизации, зародившихся на первом этапе кристаллизации, получена следующая приближенная формула:
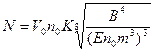 ,
,
где К » 3,5.
Расчеты на ЭВМ и эксперименты показали достаточную точность этих формул.
Величина N фактически определяет число кристаллических зерен в затвердевшей отливке, так как при t > t1 новые зерна не зарождаются. Приняв, что V0 = Npd3/6, где d — эффективный диаметр зерна, с учетом формулы для вычисления N легко найти следующее выражение для оценки величины кристаллического зерна при объемной кристаллизации:
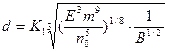 ,
,
где K1 = 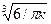 .
.
Из полученной формулы видно, что для измельчения зерна необходимо увеличить СВЦК. n0 и уменьшить линейную скорость роста т. Видно, что зерно измельчается при повышении коэффициента теплоотдачи а и снижении удельной теплоты кристаллизации L.
Рассмотренная выше схема чисто объемной кристаллизации, когда твердая фаза растет от отдельных центров во всем объеме жидкости, может быть реализована для чистых металлов при малом значении критерия Био (Bi = aR/l), т. е. при малых значениях a и R. При Bi > 0,l, как правило, объемная кристаллизация в центральных объемах сопровождается последовательным продвижением затвердевшей корки с поверхности охлаждения сплава к центру отливки. При объемной кристаллизации величина максимального переохлаждения ΔTmах невелика (для алюминия не более 1—5°С, для железа — до 40 °С). Длительность первого этапа (t £ t1) составляет относительно небольшую долю от общего времени затвердевания. Как правило, мы измеряем термопарой температуру сплава на втором этапе затвердевания, когда переохлаждение (ΔT < ΔT1) очень мало. При этом температура, фиксируемая термопарой, близка к Ткр.
Изложенная теория показывает, какое большое значение для управления формированием кристаллического строения отливки кроме теплового режима имеет целенаправленное воздействие на кристаллизационные параметры п и v.
ГЛАВА 2.5. ПЕРЕРАСПРЕДЕЛЕНИЕ ПРИМЕСЕЙ НА ФРОНТЕ КРИСТАЛЛИЗАЦИИ СПЛАВОВ. ВНУТРИКРИСТАЛЛИЧЕСКАЯ ЛИКВАЦИЯ
При кристаллизации сплавов концентрации компонентов в фазах, находящихся в равновесии, как правило, различаются. Отношение концентраций компонента в находящихся в равновесии твердой и жидкой фазах k0 = CS/CL. называется равновесным коэффициентом распределения. На рис. 2.18, а приведен участок диаграммы состояния сплава, температура ликвидуса которого уменьшается с увеличением концентрации легирующего компонента В. Очевидно, что в этом случае коэффициент распределения k0 < 1 (обычно 0,2 £ k0 £ 0,6).
При повышении температуры ликвидуса (рис. 2.18, б) коэффициент распределения k0 > 1. Обычно в этом случае k0 < 3. Однако, например, в системе Ge — В k0 достигает 15.
Дальнейшее изучение кристаллизации будем проводить при k0 < 1, так как эта величина является характерной для большинства литейных сплавов.
Рассмотрим несколько частных случаев.
1. Скорость роста кристалла настолько мала, что диффузия успевает выравнять концентрации вещества по объему твердой и жидкой фаз. При этом концентрации будут соответствовать равновесным значениям, определенным для данной температуры по диаграмме состояния. На рис. 2.19 показано распределение концентрации по длине твердой и жидкой фаз. Ось ординат, которая
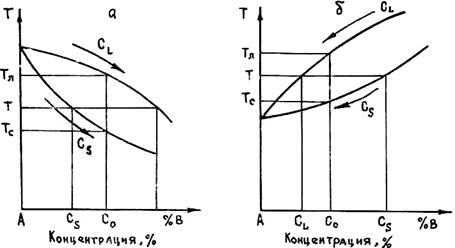
Рис. 2.18. Диаграммы состояния сплавов с понижением (а) и повышением (б) температуры ликвидуса в зависимости от роста концентрации легирующего компонента
соответствует концентрации, является подвижной и совпадает с положением фронта кристаллизации. В этом случае k0 = СS/СL.
2. Ситуация, более приближенная к реальной, имеет место, когда в твердой фазе при данной скорости роста кристалла не происходит диффузионного перераспределения вещества (Дтв = 0). Однако в жидкости, вследствие интенсивного перемешивания расплава, концентрации выравниваются. Так как Дтв = 0, то состав твердой фазы, образовавшийся на предыдущих стадиях, остается неизменным. Так как при k0 < 1 по мере кристаллизации CS увеличивается (см. рис. 2.18, а), то концентрация вещества будет возрастать по длине твердой фазы, оставаясь на фронте кристаллизации равной соответствующему значению, определенному по диаграмме состояния. Распределение концентраций по длине фаз в рассматриваемом случае приведено на рис. 2.20. Так как концентрация жидкости cl больше концентрации твердой фазы на фронте кристаллизации CS, то при затвердевании жидкости на фронте кристаллизации выделяется легирующий компонент. Например, если за время dt затвердевает dm жидкости, то количество выделившегося вещества равно (Cl—CS)dm. В данном случае это вещество равномерно распределяется по объему жидкости.
Явление, связанное с неравномерным распределением концентрации вещества, называется ликвацией. В рассматриваемом случае наблюдается максимальная ликвация в твердой фазе, называемая внутрикристаллической ликвацией. При этом внутренние слои сферического
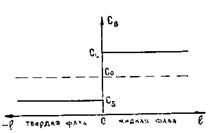
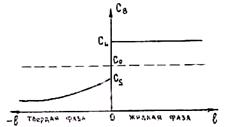
Рис. 2.19. Распределение концентрации по длине Рис. 2.20. Распределение концентраций по
жидкой и твердой фаз при малой скорости длине фаз при Дтв = 0 и Дж ® ¥
кристаллизации (k0 < 1)
кристаллита будут иметь меньшую концентрацию легирующего компонента, чем поверхностные (при слои сферического k0 < 1). Среднее содержание вещества в твердой фазе будет всегда меньше определенного по диаграмме состояния, а в жидкости, наоборот, средняя концентрация превысит равновесные значения. Это приведет к смещению влево линий солидуса на диаграмме состояния. Положение равновесной и неравновесной линии солидуса показано на рис. 2.21. Неравновесная линия солидуса отвечает средним значениям концентрации компонента в твердой фазе. Из рис. 2.21 видно, что при равновесной температуре солидуса Тсол. р часть жидкости остается незатвердевшей. Массовую долю этой жидкости можно найти по правилу рычага. Она равна отношению отрезков ae/ab. Полное затвердевание жидкости произойдет при более низкой температуре неравновесного солидуса Tсол.н.р, которая ниже равновесного значения на величину отрезка ΔT = ес.
Для выравнивания концентрации по объему кристаллитов применяют гомогенизационный отжиг, т. е. выдержку сплава при повышенных температурах, при которых значение коэффициента диффузии Дтв достаточно для диффузионного относительно быстрого выравнивания концентраций. Так как при перемешивании жидкости вещество, выделившееся на фронте затвердевания, отводится и равномерно распределяется по ее объему, то в этом случае k0 = СS/СL.
3. При переходе к более высоким скоростям роста, которые обычно имеют место, реализуется диффузионно контролируемый режим кристаллизации. Перенос вещества с фронта кристаллизации в объем жидкости осуществляется путем диффузии, и распределено оно в ней неравномерно. Примесь, выделяющаяся при образовании твердой фазы (k0 < 1), накапливается у движущегося фронта кристаллизации, и ее концентрация на фронте СL0 превышает концентрацию в объеме СL (рис. 2.22). В данном случае k0 = CS/CL0.
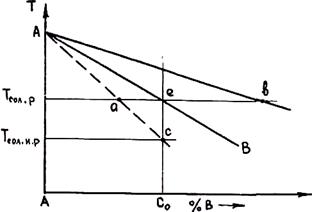
Рис. 2.21. Положение равновесной и неравновесной линий солидуса на диаграмме состояния сплава
В процессе перемещения фронта кристаллизации концентрация СL0 повышается, потому что не вся примесь отводится в расплав. Это приводит к увеличению концентрации растворенного вещества как в жидкости, так и в твердой фазе. При некотором положении фронта кристаллизации концентрация в твердой фазе достигает С0, а в расплаве на фронте — СL0 = С0/k0. Соответствующее этому моменту распределение концентраций показано на рис. 2.23.
В этих условиях можно написать следующее уравнение баланса вещества для фронта кристаллизации:
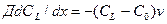 .
.
Здесь выражение в левой части определяет количество вещества, отведенного от фронта в расплав, а выражение в правой части — количество вещества, выделившегося при кристаллизации. Проинтегрировав это уравнение при начальных условиях x = 0, CL =C0/k , получаем выражение, описывающее распределение концентрации примеси в расплаве вблизи фронта кристаллизации:
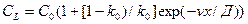 . (2.5)
. (2.5)
На некотором расстоянии х = d cl практически равно С0. Величина d называется толщиной диффузионного пограничного слоя.
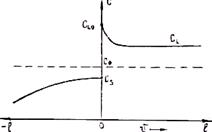

Рис. 2.22. Распределение примеси при диффузионно Рис. 2.23. Распределение концентраций в
контролируемой кристаллизации момент достижения их предельных значений
на фронте кристаллизации
Она зависит от интенсивности движения жидкости. С увеличением скорости движения жидкости, особенно при перемешивании, d быстро убывает. Если жидкость покоится, то d = Д/(v× k0).
ГЛАВА 2.6. КОНЦЕНТРАЦИОННОЕ ПЕРЕОХЛАЖДЕНИЕ. УСЛОВИЕ УСТОЙЧИВОСТИ ПЛОСКОГО ФРОНТА КРИСТАЛЛИЗАЦИИ СПЛАВОВ
При достаточно больших скоростях кристаллизации концентрация легирующего компонента перед фронтом кристаллизации распределена неравномерно. Распределение концентрации в расплаве описывает формула (2.5), из которой видно, что по мере удаления от фронта cl более или менее быстро убывает.
Как видно из диаграммы состояния (см. рис. 2.18, а), температура ликвидуса с ростом концентрации легирующего компонента уменьшается. Если принять линии ликвидуса и солидуса прямыми (рис. 2.24), то для зависимости температуры ликвидуса от концентрации компонента cl справедливо следующее уравнение:
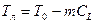 ,
,
где Т0 — температура плавления металла, являющегося основой сплава; m = tg a — тангенс угла наклона прямой ликвидуса к оси концентрации компонента В.
С учетом зависимости СL(х) получаем уравнение
 ,
,
из которого следует, что температура ликвидуса сплава перед фронтом кристаллизации по мере удаления от него увеличивается, достигая при х = d значения Tл, отвечающего исходной концентрации сплава С0. Температура ликвидуса сплава на фронте кристаллизации (СL0 = С0/k0) равна Tф = T0 – mC0/k0. Схема распределения температуры ликвидуса в расплаве перед фронтом кристаллизации приведена на рис. 2.25. При охлаждении сплава в нем возникает температурный градиент через твердую фазу, равный на фронте кристаллизации GR = (dT/dx)x=0. Примем линейное распределение температуры сплава Т (х) в пределах пограничного слоя d. При этом T(x) = Tф + GR×x. Следует отметить, что, приняв Т (х = 0) = Тф, мы пренебрегаем термическим переохлаждениемΔT.
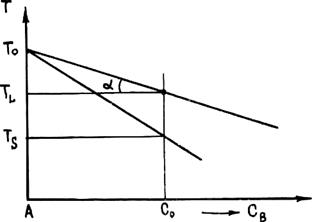
Рис. 2.24. Схема к выводу зависимости Тл(СL)
На рис. 2.25 линия 1 отвечает кривой изменения температуры ликвидуса, а линия 2 — распределению температуры сплава перед фронтом кристаллизации. В пределах слоя 0 £ х £ dп температура сплава ниже температуры ликвидуса, т. е. сплав является переохлажденным. Это переохлаждение обусловлено изменением состава сплава в процессе кристаллизации и называется концентрационным переохлаждением ΔТк в отличие от рассмотренного в гл. 2.1 термического переохлаждения ΔT, обусловленного падением температуры сплава за счет теплоотвода ниже температуры ликвидуса на фронте кристаллизации или в какой-либо части объема жидкости.
Наличие концентрационного переохлаждения создает условия для нарушения плоского характера фронта кристаллизации. На фронте кристаллизации всегда имеются шероховатости с выступами микроскопических размеров. Вершины этих выступов, находящиеся перед фронтом кристаллизации, попадают в зону концентрационного переохлаждения, что существенно повышает скорость их продвижения в глубь расплава, так как с увеличением переохлаждения любой природы линейная скорость роста кристалла возрастает. Поэтому скорость продвижения в глубь расплава вершин выступов больше, чем оснований, находящихся на фронте, где ΔTк = 0 (см. рис. 2.25). В результате высота выступов увеличивается и плоский фронт кристаллизации нарушается.
Дата добавления: 2014-12-09; просмотров: 1730;