Законодательная практика Европейского Союза (ЕС) и США
При выведении на рынок новых лекарственных средств предусмотрена процедура выдачи разрешения, состоящего из двух самостоятельных пакетов: испытаний и регистрации нового препарата и лицензирования его производства. В настоящее время международная фармацевтическая практика оперирует следующими терминами, характеризующими указанную процедуру:
Product License = Marketing Authorization – торговая лицензия (соответствует российскому регистрационному удостоверению на лекарственное средство);
Manufacturer’s License = Manufacturing Authorization – лицензия на производство лекарственных средств.
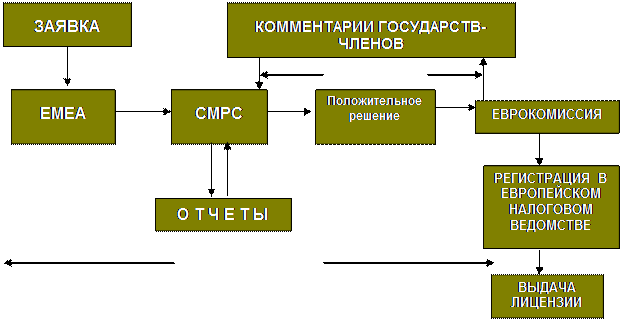
В ЕС действует централизованная процедура выдачи торговой лицензии, представленная на рис.1.
Менее распространена в ЕС занимающая примерно 1 год так называемая децентрализованная выдача лицензии, состоящая в регистрации лекарственного средства в национальном регуляторном и налоговом органе с последующим прохождением через процедуру взаимного признания в государствах-членах, а затем через Еврокомиссию (исполнительный орган ЕС).
В ЕС практикуется также выдача лицензии на лекарственное средство для клинических исследований (Investigational Medicinal Products Authorization), а в UK – лицензии на оптовую торговлю (импорт) лекарственных средств (Wholesale Dealer’s (Import) License).
Выдачей лицензий в Соединенном Королевстве ведает исполнительный регламентирующий орган Департамента (Министерства) здравоохранения – Агентство по лекарственным средствам, изделиям медицинского назначения и медицинской технике (MHRA – Medicines and Healthcare products Regulatory Agency).
 |
Дней
|
EMEA– Европейское агентство экспертизы лекарственных средств
CMPC – Комитет ЕМЕА по лекарственным средствам для человека
Рис.1 Схема процедуры централизованной выдачи торговой лицензии в Евросоюзе
В США производитель лекарственных средств, наряду с получением в FDA лицензии на производство, должен также пройти процедуру обязательной регистрации в Минюсте по форме DEA 225A в соответствии с Актом по контролируемым веществам от 1970 г., где осуществляется проверка его чистоты перед законом. Особенно жестко это требование стало предъявляться после 11 сентября 2001 г. в связи с принятием программ борьбы с биотерроризмом LEADERS (Lightweight Epidemiology Advanced Detection and Emergency Response System) и RSVP (Rapid Syndrome Validation Project).
Наиболее полно регуляторные аспекты обращения лекарственных средств в ЕС представлены в базе данных Eudralex Collection (http://pharmacos.eudra.org), включающей 9 томов информации следующего содержания:
Т.1 – Фармацевтическое законодательство
Лекарственные средства для человека
Т.2 – Инструкции для заявителей
Лекарственные средства для человека
Т.3 – Руководства
Лекарственные средства для человека и ветеринарные ЛС
Т.4 – GMP. Лекарственные средства для человека и ветеринарные ЛС
Т.5 – Фармацевтическое законодательство
Ветеринарные лекарственные средства
Т.6 – Инструкции для заявителей
Ветеринарные лекарственные средства
Т.7 – Руководства
Ветеринарные лекарственные средства
Т.8 – Максимальный остаточный уровень
Ветеринарные лекарственные средства
Т.9 – Безопасность лекарственных средств
Лекарственные средства для человека и ветеринарные ЛС
Указанный Интернет-сайт постоянно обновляется и позволяет осуществлять эффективный мониторинг нормативно-технической и нормативно-правовой информации в сфере обращения лекарственных средств на 11 языках стран – членов ЕС. Заинтересованные лица в РФ могут ознакомиться с сопоставленным англо-русским переводом основополагающей Директивы 2001/83/ЕС Европейского Парламента и Совета ЕС от 6 ноября 2001 г. «О своде законов Сообщества в отношении лекарственных препаратов для человека», утвердившей порядок регистрации, производства, продажи и использования лекарственных средств, на сайте http://www.webapteka.ru.
Дата добавления: 2016-09-20; просмотров: 1021;