Глава V. Реакционная способность комплексов.
Условно химические реакции комплексов подразделяют на обменные, окислительно-восстановительные, изомеризации и координированных лигандов.
5.1. Обменные реакции. Взаимное влияние лигандов.
Первичная диссоциация комплексов на внутреннюю и внешнюю сферу определяет протекание реакций обмена внешнесферных ионов:
[MLn]Xm + mNaY = [MLn]Ym + mNaX.
Компоненты внутренней сферы комплексов также могут участвовать в обменных процессах с участием как лигандов, так и комплексообразователя. Для характеристики реакций замещения лигандов или центрального иона металла используют обозначения и терминологию, предложенную К. Ингольдом для реакций органических соединений, нуклеофильного SN и электрофильного SE замещения:
[MLnX]z + Y = [MLnY]z +X SN
[MLn]z + M’= [M’Ln]z + M SE.
По механизму реакции замещения разделяют на ассоциативный (SN1 и SE1) и диссоциативный (SN2 и SE2), различающиеся переходным состоянием с увеличенным и уменьшенным координационным числом:
Отнесение механизма реакции к ассоциативному или диссоциативному является трудно экспериментально достижимая задача идентификации интермедиата с уменьшенным или увеличенным координационным числом. В связи с этим, часто о механизме реакции судят на основании косвенных данных о влиянии концентрации реагентов на скорость реакции, изменении геометрического строения продукта реакции и др.
Для характеристики скорости реакций замещения лигандов комплексов, нобелевский лауреат 1983 г. Г. Таубе предложил использовать термины «лабильный» и «инертный» в зависимости от времени протекания реакции замещения лигандов менее или более 1 минуты. Термины лабильный или инертный являются характеристикой кинетики реакций замещения лигандов и их не следует путать с термодинамическими характеристиками об устойчивости или нестойкости комплексов.
Лабильность или инертность комплексов зависит от природы иона комплексообразователя и лигандов. В согласии с теорией поля лигандов:
1. Октаэдрические комплексы 3d переходных металлов с распределением валентных (n-1)d электронов на s*(eg) разрыхляющих МО лабильны.
[CoII(CN)6]4-(t2g6eg1) + H2O ® [Co(CN)5H2O]3- + CN-.
Причем, чем меньше величина энергии стабилизации кристаллическим полем комплекса, тем его лабильность больше.
2. Октаэдрические комплексы 3d переходных металлов со свободными s* разрыхляющими eg орбиталями и равномерным распределением валентных (n-1)d электронов по t2g орбиталям (t2g3, t2g6) инертны.
[CoIII(CN)6]3-(t2g6eg0) + H2O =
[CrIII(CN)6]3-(t2g3eg0) + H2O =
3. Плоско-квадратные и октаэдрические 4d и 5d переходных металлов, не имеющие электронов на s* разрыхляющих МО инертны.
[PtII(NH3)4]2+ + H2O =
[RhIII(NH3)5Cl]2+ + H2O =
Влияние природы лигандов на скорость реакций замещения лигандов рассматривается в рамках модели «взаимного влияния лигандов». Частным случаем модели взаимного влияния лигандов является, сформулированная в 1926 г. И.И. Черняевым концепция транс-влияния - «лабильность лиганда в комплексе зависит от природы транс-расположенного лиганда» - и предложить ряд транс-влияния лигандов: CO, CN-, C2H4 > PR3, H- > CH3-, SC(NH2)2 > C6H5-, NO2-, I-, SCN- > Br-, Cl- > py, NH3, OH-, H2O.
Концепция транс-влияния позволила обосновать эмпирические правила:
1. Правило Пейроне – при действии аммиака или аминов на тетрахлоплатинат(II) калия получается всегда дихлодиаминплатина цис-конфигурации:
[PtCl4]2- + 2NH3 = цис-[Pt(NH3)2Cl2] + 2Cl-.
Поскольку реакция протекает в две стадии и хлоридный лиганд обладает большим транс-влиянием, то замещение второго хлоридного лиганда на аммиак происходит с образованием цис-[Pt(NH3)2Cl2]:
[PtCl4]2- + NH3 = [Pt(NH3)Cl3]-
[Pt(NH3)Cl3]- + NH3 = цис-[Pt(NH3)2Cl2].
2. Правило Иергенсена – при действии хлороводородной кислоты на хлорид тетраммина платины(II) или подобные соединения получается дихлородиамминплатина транс-конфигурации:
[Pt(NH3)4]2+ + 2HCl = транс-[Pt(NH3)2Cl2] + 2NH4Cl.
В соответствии с рядом транс-влияния лигандов, замещение второй молекулы аммиака на хлоридный лиганд приводит к образованию транс-[Pt(NH3)2Cl2].
3. Тиомочевинная реакция Курнакова – различные продукты реакции тиомочевины с геометрическими изомерами транс-[Pt(NH3)2Cl2] и цис-[Pt(NH3)2Cl2]:
транс-[Pt(NH3)2Cl2] + 2Thio = транс-[Pt(NH3)2(Thio)2]2+
цис-[Pt(NH3)2Cl2] + 4Thio = [Pt(Thio)4]2+ + 2Cl- + 2NH3.
Различный характер продуктов реакции связан с высоким транс-влиянием тиомочевины. Первой стадией реакций является замещение тиомочевинной хлоридных лигандов с образованием транс- и цис- [Pt(NH3)2(Thio)2]2+:
транс-[Pt(NH3)2Cl2] + 2Thio = транс-[Pt(NH3)2(Thio)2]2+
цис-[Pt(NH3)2Cl2] + 2Thio = цис-[Pt(NH3)2(Thio)2]2+.
В цис-[Pt(NH3)2(Thio)2]2+ две молекулы аммиака находящиеся в транс положении к тиомочевине подвергаются дальнейшему замещению, что и приводит к образованию [Pt(Thio)4]2+:
цис-[Pt(NH3)2(Thio)2]2+ + 2Thio = [Pt(Thio)4]2+ + 2NH3.
В транс-[Pt(NH3)2(Thio)2]2+ две молекулы аммиака с малым транс-влиянием расположены в транс положении друг к другу и поэтому не замещаются тиомочевинной.
Закономерности транс-влияния широко используются при выборе пути синтеза комплексных соединений. Примером использования транс-влияния синтез, осуществленный Гельманом и Эссеном, трех изомеров:
Синтез первого изомера был проведен по реакциям:
транс-[PtPyNH3Cl2] + AgNO3 + H2O = [PtPyNH3H2Ocl]NO3 + AgCl¯
[PtPyNH3H2Ocl2]NO3 + KBr = [PtPyNH3BrCl] + KNO3 + H2O.
Для синтеза второго изомера в качестве исходного комплекса использовали K[PtNH3Cl3] и проводили реакции с KBr и пиридином:
K[PtNH3Cl3] + KBr = K[PtNH3Cl2Br] + KCl
K[PtNH3Cl2Br] + Py = [PtPyNH3BrCl] + KCl.
Третий изомер был получен из K[PtPyCl3] в реультате взаимодействия с KBr и затем аммиаком:
K[PtPyCl3] + KBr = K[PtPyCl2Br] + KCl
K[PtPyCl2Br] + NH3 = [PtPyNH3BrCl] + KCl.
Закономерности транс-влияния были открыты И.И. Черняевым при изучении реакций замещения лигандов в плоско-квадратных комплексах платины(II). В дальнейшем было показано, что транс-влияние лигандов проявляется и в комплексах других металлов (Pt(IV), Pd(II), Co(III), Cr(III), Rh(III), Ir(III)) и другого геометрического строения. Правда, ряды транс-влияния лигандов для разных металлов несколько различаются.
Слеудует отметить, что транс-влияние является кинетическим эффектом – чем большим транс-влиянием обладает данный лиганд, тем с большей скоростью замещается другой лиганд, находящийся по отношению к нему в транс-положении.
Наряду с кинетическим эффектом транс-влияния, в середине XX века А.А. Гринбергом и Ю.Н. Кукушкиным установлено зависимость транс-влияния лиганда L от лиганда, находящегося в цис-положении к L. Так, исследование скорости реакции замещения Cl- аммиаком в комплексах платины(II):
[PtCl4]2- + NH3 = [PtNH3Cl3]- + Cl- K = 0.42.104 л/моль.с
[PtNH3Cl3]- + NH3 = цис-[Pt(NH3)2Cl2] + Cl- K = 1.14.104 л/моль.с
транс-[Pt(NH3)2Cl2] + NH3 = [Pt(NH3)3Cl]+ + Cl- K = 2.90.104 л/моль.с
показало, что наличие в цис-положении к замещаемому хлоридному лиганду одной и двух молекул аммиака приводит к последовательному увеличению скорости реакции. Этот кинетический эффект получил название цис-влияние. В настоящее время оба кинетических эффекта влияния природы лигандов на скорость реакций замещения лигандов (транс- и цис-влияние) объеденены в общей концепции взаимного влияния лигандов.
Теоретическое обоснование эффекта взаимного влияния лигандов тесно связано с развитием представлений о химической связи в комплексных соединениях. В 30-х годах XX века А.А. Гринбергом и Б.В. Некрасовым транс-влияние рассмотрено в рамках поляризационной модели:
1. Транс-влияние характерно для комплексов, центральный ион металла которых обладает большой поляризуемостью.
2. Транс-активность лигандов определяется величиной энергии взаимной поляризации лиганда и иона металла. Для данного иона металла транс-влияние лиганда определяется его поляризуемостью и расстоянием от центрального иона.
Поляризационная модель согласуется с экспериментальными данными для комплексов с простыми анионными лигандами, например, галогенид-ионами.
В 1943 г. А.А. Гринберг выдвинул предположение, что транс-активность лигандов связана с их восстановительными свойствами. Смещение электронной плотности от транс-активного лиганда к металлу уменьшает эффективный заряд иона металла, что приводит к ослаблению химической связи с транс-расположенным лигандом.
Развитие представлений о транс-влиянии связано с большой транс-активностью лигандов на основе ненасыщенных органических молекул, подобных этилену в [Pt(C2H4)Cl3]-. По мнению Чатта и Оргела, это связано с p дативным взаимодействием таких лигандов с металлом и ассоциативным механизмом реакций замещения транс-расположенных лигандов:
Координация к иону металла атакующего лиганда Z приводит к образованию пятикоординационного тригонально-бипирамидального интермедиата с последующим быстрым отщеплением уходящего лиганда Х. Образованию такого интермедиата способствует p дативное лиганд-металл взаимодействие лиганда Y (рис. 5.1), уменьшающая электронную плотность металла и снижающая энергию активации переходного состояния с последущим быстрым замещением лиганда Х.
Рис. 5.1. Смещение электронной плотности металла при p дативной химической связи лиганд-металл.
Наряду с p акцепторными (C2H4, CN-, CO…) лигандами, образующими дативную лиганд-металл химическую связь, высоким транс-влиянием обладают и s донорные лиганды: H-, CH3-, C2H5-… Транс-влияние таких лигандов определяется донорно-акцепторным взаимодействием лиганда Х с металлом, понижающего его электронную плотность и ослабляющего связь металла с уходящим лигандом Y (рис. 5.2).
Рис. 5.2. Транс-активность s донорных Y лигандов.
Таким образом, положение лигандов в ряду транс-активности определятся совместным действием s донорных и p свойств лигандов - s донорные и p акцепторные свойства лиганда усиливают его транс-влияние, тогда как p донорные – ослабляют. Какая из этих составляющих лиганд-металл взаимодействия преобладает в транс-влиянии судят на основании квантово-химических расчетов электроного строения переходного состояния реакции.
5.2. Окислительно-восстановительные реакции комплексов.
Количественной характеристикой окислительно-восстановительных (Ox-Red) свойств комплексов является величина потенциала, характеризующего равновесие между окисленной (Ox) и восстановленной (Red) формами:
Ox + ze- « Red E(Ox/Red)
По сравнению с положением металла в ряду напряжений, определяемого величиной стандартного потенциала равновесия:
[M(H2O)n]z+ + ze- « M + nH2O E0([M(H2O)n]z+/M)
значения стандартных потенциалов комплексов значительно изменяются в зависимости от величины констант нестойкости комплексов с разным лигандным окружением в соответствии с уравнением:
E0(MLnz/M) = E0(M(H2O)nz+/M) + 0.059lgKн
Так, например, с уменьшением констант нестойкости комплексов серебра(I) их восстановительные свойства возрастают:
[Ag(H2O)2]+ + e- « Ag + 2H2O E0(Ag(H2O)2+/Ag) = 0.8 В
[Ag(NH3)2]+ + e- « Ag + 2NH3 E0(Ag(NH3)2+/Ag) = 0.37 В,
(Kн = 5.9.10-8)
[Ag(S2O3)2]3- + e- « Ag + 2S2O32- E0(Ag(S2O3)23-/Ag) = -0.26 В
(Kн = 1.10-18)
[Ag(CN)2]- + e- « Ag + 2CN- E0(Ag(CN)2-/Ag) = -0.44 В
(Kн=1.1.10-21)
Величина потенциала Ox-Red системы комплексов, содержащей ионы металла с различной степенью окисления:
[MLn]z + ze- « [MLn]z1
по сравнению с аквакомплексами зависит от соотношения констант нестойкости восстановленной и окисленной форм:
E0(MLnz/MLnz1) = E0(M(H2O)nP+/M(H2O)nm+) + (0.059/z)lg(Kн(Red)/Kн(Ox)).
Например, величина потенциала равновесия между аквакомплексами Fe(II) и Fe(III) увеличивается и уменьшается для комплексов с бипиридильными и цианидными лигандами:
[Fe(H2O)6]3+ + e- « [Fe(H2O)6]2+ E0(Fe(H2O)63+/Fe(H2O)62+) = 0.77 В
[Fe(bpy)3]3+ + e- = [Fe(bpy)3]2+ E0(Fe(bpy)33+/Fe(bpy)32+) = 0.90 В
Кн = 2.10-21 Кн = 1.10-23
[Fe(CN)6]3- + e- = [Fe(CN)6]4- E0(Fe(CN)63-/Fe(CN)64-) = 0.36 В
Кн = 1.10-31 Кн = 1.10-24
Изменение электронного состояния металла в результате Ox-Red реакции может приводить к изменению координационного числа иона металла. Так, 2-х электронное восстановление октаэдрических комплексов ионов металлов с 4d6 и 5d6 электронной конфигурацией сопровождается восстановительным элиминированием аксиальных лигандов с образованием плоско-квадратных комплексов 4d8 и 5d8 электронной конфигурации металла. При обратном процессе – окислительного присоединения – плоско-квадратные комплексы окисляются в октаэдрические:
[PtIV(NH3)4Cl2]2+ +2e-=[PtII(NH3)4]2++2Cl- (восстановительное элиминирование)
[PtII(NH3)4]2+ + 2Cl- + 2e- = [PtIV(NH3)4Cl2]2+ (окислительное присоединение).
Исследование механизма Ox-Red реакций комплексов, определяющего скорость их протекания, позволило нобелевскому лауреату 1983 г. Г. Таубе выдвинуть предположение о внутрисферном (мостиковом) и внешнесферном (тунельном) механизме протекания таких реакций.
Различие внутри- и внешнесферного механизма Ox-Red реакций комплексов определяется различной природой переходного состояния. При внутрисферном механизме диффузионное сближение реагентов приводит к образованию «мостикового» переходного состояния:
[CrII(H2O)6]2+ + [CoIII(CN)5Cl]3- « {(H2O)5Cr-Cl-Co(CN)5}-,
трансформация которого в продукты реакции сопровождается переносом мостикового атома или группы атомов:
{(H2O)5Cr-Cl-Co(CN)5}- « [CrIII(H2O)5Cl]2+ + [CoII(CN)5]3-.
Лимитирующей стадией, определяющей в целом скорость Ox-Red реакций комплексов при внутрисферном механизме является стадия образования мостикового интермедиата. Эффективность образования мостикового интермедита в значительной степени зависит от природы мостикового атома или группы атомов. Например, изменение природы галогена в составе пентамминовых комплексов Co(III) приводит к увеличению скорости реакций их восстановления аквакомплексом Cr(II):
[CrII(H2O)6]2+ + [CoIII(NH3)5Hal]2+ = [CrIII(H2O)5Cl]2+ + Co(II)aq + 5NH3
[Co(NH3)5F]2+ K = 2.5.105 л/моль.с, [Co(NH3)5Cl]2+ K = 6.0.105 л/моль.с, [Co(NH3)5Br]2+ K = 1.4.106 л/моль.с, [Co(NH3)5I]2+ K = 3.0.106 л/моль.с.
При внешнесферном механизме диффузионное сближение реагентов с инертным внутрисферным окружением приводит к образованию «комплекса встреч» с контактирующим лигандным окружением:
[FeII(CN)6]4- + [RuIII(bpy)3]3+ « {[FeII(CN)6]4-…[RuII(bpy)3]3+}
и последующим тунельным переносом электрона:
{[FeII(CN)6]4-…[RuII(bpy)3]3+} « [FeIII(CN)6]3- + [RuII(bpy)3]2+.
Лимитирующей стадией Ox-Red реакций комплексов при внешнесферном механизме является стадия тунельного переноса электрона в комплексе встреч. Скорость туннельного переноса электрона между компонентами комплекса встреч зависит от величины изменения равновесных расстояний DR лиганд-металл в окисленной и восстановленной формах реагентов. Величина DR в свою очередь зависит от изменения числа электронов металла на s* (eg) МО при изменении степени окисления иона металла. Так, для реакции между низкоспиновыми комплексами [FeII(CN)6]4- (t2g6eo0) и [RuIII(bpy)3]3+ (t2g5eo0) перенос электрона с образованием [FeIII(CN)6]3- (t2g5eo0) и [RuII(bpy)3]2+ (t2g6eo0) не сопровождается изменением числа электронов реагентов на eg орбиталях. Это приводит к незначительному изменению величины DR и высокой скорости туннельного переноса электрона и всей Ox-Red реакции (К ~109 л/моль.с).
При взимодействии высокоспинового [CrII(H2O)6]2+ (t2g3eg1) с [RuIII(bpy)3]3+ (t2g5eg0):
[CrII(H2O)6]2+ + [RuIII(bpy)3]3+ = [CrIII(H2O)6]3+ + [RuII(bpy)3]2+
тунельный перенос электрона в комплексе встреч с образованием [CrIII(H2O)6]3+ (t2g3eg0) и [RuII(bpy)3]2+ (t2g6eo0) сопровождается изменением электронов на eg орбиталях хрома, что определяет изменение величины DR в комплексах хрома и приводит к значительному уменьшению скорости тунельного переноса электрона и всей Ox-Red реакции (К ~106 л/моль.с).
5.3. Реакции изомеризации.
Реакции изомеризации протекают как в расторах, так в твердой фазе. Растворитель существенно влияет на механизм и энергетику процесса. Так, в полярных растворителях равновесие между цис- и транс- изомерами комплексов практически нацело смещено в сторону более полярного цис-изомера. Например, при концентрации на водяной бане водного раствора зеленого транс-[CoEn2Cl2]Cl изомера выделяются фиолетовые кристаллы цис-[CoEn2Cl2]Cl изомера. В свою очередь эта фиолетовая соль переходит в зеленый транс-[CoEn2Cl2]Cl изомер упариванием солянокислого раствора. При изомеризации в твердой фазе важную роль играет различие в энергии кристаллической решетке изомеров. В общем случае изомеризация в твердой фазе протекает легче, чем в растворе.
Термодинамическое обоснование изомеризации цис-[PtA2X2] в транс–[PtA2X2] (А – амин, X – галогены) было получено И.И. Черняевым, установившим, что полная энергия транс-изомеров меньше энергии цис-изомеров.
На основании взаимосвязи транс-влияния лигандов в составе внутренней сферы [PtA2X2] комплексов Ю.Н. Кукушкин сформулировал общее правило протекания изомеризации в плоско-квадратных комплексах Pt(II) – процесс изомеризации идет в направлении образования комплекса, в котором амин располагается напротив лиганда с наименьшим транс-влиянием. Правило изомеризации комплексов Pt(II) распространяется не только на аминные, но и фосфиновые комплексы. Например, транс-[Pt(PPh3)(R2S)Cl2] при нагревании переходит в цис-изомер, при этом трифенилфосфиновый PPh3 и тиоэфирный R2S лиганды занимают транс-положение к хлоридным лигандам с наименьшим транс-влиянием.
Предполагается, что твердофазная изомеризация плоско-квадратных комплексов протекает через переходное состояние тетраэдрической структуры. В тоже время, для реакций геометрической изомеризации в растворах предполагается диссоциативный механизм с образованием пятикоординационного интермедиата.
Наряду с геометрической, реакциям изомеризации как в твердой фазе, так и в растворах подвергаются и другие типы изомеров. Например, в твердой фазе при нагревании нитрито-изомеры в результате внутримолекулярной перегруппировки эффективно переходят в нитро-изомеры:
Подобный внутримолекулярный механизм приводит к твердофазной тиоцианат-изотиоцианат изомеризации:
[(bpy)2Pd(SCN)2] ® [(bpy)2Pd(NCS)2].
В тоже время, в растворах реакции связевой изомеризации, подобно реакциям геометрической изомеризации, могут протекать по диссоциативному механизму, приводящему к перекоординации SCN- в NCS- лиганд.
Эфективность реакций рацемизация оптических изомеров комплексов в растворе в значительной степени определяется их лабильностью. Так, в отличие от инертного [CoEn3]3+, лабильный (+) [CoEn2Cl2]+ легко теряет свою оптическую активность в водных растворах в результате рацемизации и образования смеси оптических изомеров.
5.4. Реакции координированных лигандов.
Изменение электронного строения лиганда при координации к иону металла неизбежно приводит к изменению его реакционной способности. Еще до появления самого понятия координированного лиганда М.П. Шютценберг в 1872-1874 гг. установил, что гидролиз, координированного к платине(II) PCl3, приводит к образованию координированной фосфористой кислоты:
[Pt(PCl3)2Cl2] + 6H2O = [Pt(P(OH)3)2Cl2] + 6HCl.
В отличие от свободной двухосновной фосфористой кислоты (рис. 5.3) с сильными восстановительными свойствами, координированная фосфористая кислота трехосновна и обладает слабыми восстановительными свойствами.
Рис. 5.3. Строение свободной и координированной фосфористой кислоты.
К.А. Гофман, Г. Бугге (1908 г.) показали превращение координированных нитрилов RCN в ацетамид:
RCN + H2O = RC(O)NH2,
а Л.А. Чугаев в 1921 г. осуществил гидролиз внутрисферного цианатного лиганда с образованием координированной карбаминовой кислоты:
цис-[Pt(NH3)2Cl(NCO)] + H2O ® цис-[Pt(NH3)2Cl(NHCOOH].
Приведенные факты показывают глубинную историю исследований реакций координированных лигандов. В тоже время, интенсивные исследования этих реакций практически начались в 60-х годах XX века. В 1968-1970 гг. были опубликованы первые книги, посвященные изменению реакционной способности координированных лигандов – М.М. Джонс «Реакционная способность лигандов и катализ» и Дж. Кендлин, К. Тейлор, Д. Томпсон «Реакции координационных соединений переходных металлов». Бурное развитие этого направления координационной химии в настоящее время связано с несомненной определяющей ролью изменения реакционной способности лигандов при координации в металлокомплексном катализе.
Донорно-акцепторное взаимодействие с металлом приводит к смещению электронной плотности от донорного атома лиганда к металлу, увеличению степени ионности его связи с соседними атомами лиганда (рис. 5.4) и увеличению эффективности процесса ее электролитической диссоциации.
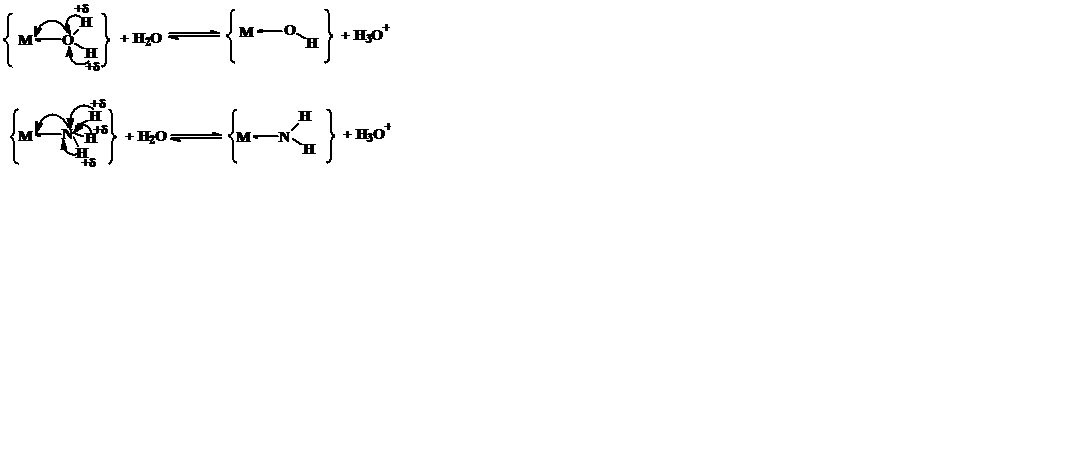
Рис. 5.4. Увеличение кислотных свойств координированных H2O и NH3.
Это определяет увеличение кислотных свойств координированных аминов, воды и других протон содержащих лигандов:
2H2O « H3O+ + OH- K = 1.8.10-16
[Pt(NH3)5H2O]4+ + H2O « [Pt(NH3)5OH]3+ + H3O+ K = 3.10-10
NH3 + H2O « H3O+ + NH2- K < 10-25
[Au(NH3)4]3+ + H2O « [Au(NH3)3NH2]2+ + H3O+ K ~ 10-4
Кислотная диссоциация координированной воды может приводить к перестройке координационной сферы комплекса:
или оляции с образованием мостиковых полиядерных комплексов:
2[LnM(H2O)]z + 2H2O « [LnM(µ-OH)]2z1 + 2H2O+
2[LnM(H2O)]z + 4H2O « [LnM(µ-O)]2z2 + 4H2O+
Депротонирование координированного лиганда приводит к образованию на нем электронной пары и усилению электронно-донорных свойств лиганда, что определяет образование хлораминов и взаимодействие амидо комплексов Pt(IV) с ионами серебра:
[Pt(NH3)4Cl2]2+ + Cl2 = [Pt(NH3)3(NH2Cl)Cl]+ + HCl
[Pt(NH3)3(NH2)Cl2]+ + Ag+ = [Pt(NH3)3(NH2Ag)Cl2]2+.
Депротонирование внутрисферных нитрилов с образованием карбоанионов:
[Co(NH3)5(NCCH2R)]3+ + OH- = [Co(NH3)5(NCCHR)]2+ + H2O
сопровождается внутримолекулярным окислительно-восстановительным процессом с образованием Co(II) и димерного нитрила:
2[Co(NH3)5(NCCHR)]2+ ® 2Со(II)aq + 10NH3 + (NCCHR)2.
Координация лиганда к иону металла сопровождатся увеличением его электрофильности и склонности к взаимодействию с нуклеофилами. Это отражается в усилении гидролиза координированных лигандов по сравнению со свободными. Например, тиомочевинные комплексы платиновых металлов при нагревании в водных растворах разлагаются с выделением сульфидов, что связано с усилением гидролиза внутрисферной тиомочевины:
и последующего разложения нестойкого продукта гидролиза.
Окисление донорных атомов S – лигандов «мягких» оснований (R3PS, R3Pse, R3Pte…) во внутренней сфере комплексов с «жесткими» ионами металлов (Nb(V), Ta(V)… ) приводит к образованию более устойчивого комплекса «жесткого» иона металла с «жестким» донорным атомом O:
2[Ta(SPR3)F5] + O2 = 2[Ta(OPR3)F5] + 2S
Изменение реакционной способности лиганда при координации позволяет на основе реакций присоединения и конденсации получать комплексы с новыми макроциклическими лигандами:
Перераспределение электронной плотности поликарбоновых кетокислот при координации вызывает их каталитическое декарбоксилирование:
Одной из ключевых реакций в механизме металлокомплексного катализа получения органических соединений являются реакции внедрения или миграции лигандов в металлоорганических комплексах, Такие реакции включают координацию лиганда к металлу и последующее взаимодействие с соседним лигандом c к образованием нового органического соединения:
В качестве лигандов Х и Y могут выступать различные химические частицы: X = H-, R-, OR-; Y = CO, R2C=CR2, RCº CR, RCHO, RCN, SO2, O2.
Механизм реакций внедрения установлен в 1957 г. Коффилдом с помощью меченной 13С окиси углерода при ее взаимодействие с пентакарбонилметилмарганцем:
[(CH3)Mn(CO)5] + *CO = [(CH3CO)Mn(*CO)(CO)4].
Показано, что реакция сопровождается образованием семикоординационного интермедиата с последующей миграцией метильной группы:
Приведенные примеры реакций координированных лигандов показывает как их многообразие, так и практическое значение для металлокомплексного катализа и других областей прикладной координационной химии.
Контрольные вопросы.
1. Что означают символы SN1, SE1, SN2, SE2, используемые при описании реакций замещения комплексных соединений?
2. Какие комплексы называют «лабильными» и какие «инертными»?
3. Какие из комплексов: [Pt(NH3)4Cl2]2+, [Cr(NH3)4Cl2]+, [Co(NH3)4Cl2]+, [Rh(NH3)4Cl2] являются «лабильными» и какие «инертными»?
4. В чем состоит явление транс-влияния лигандов? Является ли транс-влияние лигандов термодинамическим или кинетическим эффектом?
5. В чем заключаются правила Пейроне и Иергенсена и как они согласуется с эффектом транс-влияния лигандов?
6. На чем основан метод определения геометрических изомеров комплексов Pt(II) с помощью «тиомочевинной реакции Курнакова»?
7. Обоснуйте высокое транс-влияние лигандов: CO, CN-, C2H4, CH3-, H-.
8. Используя концепцию транс-влияния лигандов, предложите схемы синтеза следующих комплексов исходя из K2[PtCl4]: а) цис-[PtPyCl2], б) транс-[Pt(NH3)2I2], в) цис-[Pt(NH3)PyCl2], г) транс-[Pt(NH3)PyCl2].
9. В чем заключается эффект цис-влияния?
10. Как и почему устойчивость комплекса влияет на величину потенциала Ox-Red равновесия [MLn]m + ze- « M + nLp ?
11. Какой цианидный комплекс Fe(II) или Fe(III) более устойчив, если величина стандартного потенциала равновесия [Fe(CN)6]4- + e- « [Fe(CN)6]4- составляет 0.36 В по сравнению с 0.77 В для равновесия [Fe(H2O)6]3+ + e- « [Fe(H2O)6]2+ ?
12. В чем заключается между внутрисферным и внешнесферным механизмом Ox-Red реакций комплексов?
13. В чем заключается правило «тран-цис изомеризации» плоско-квадратных комплексов Pt(II) ?
14. В чем причина подкисления водного раствора при растворении [Pt(NH6)6]Cl ?
Дата добавления: 2016-01-03; просмотров: 3606;