Глава IV. Модели химической связи в комплексных соединениях.
4.1. Ионная модель.
Формирование в 1893 г. и развитие координационной теории А. Вернера тесно связано с представлениями о характере химической связи в соединениях. В 1916 г. В. Коссель (1916 г.) рассмотрел химическую связь как результат электростатического взаимодействия ионов, тогда как Дж. Люис – как результат образования общих электронных пар при взаимодействии атомов химических элементов.
В соответствии с представлениями В. Косселя, устойчивому состоянию системы соответствует максимальная координация анионов лигандов вокруг катиона комплексообразователя с минимальным отталкиванием между лигандами. Так, образование комплексного иона [Ag(I-)2]- соответствует минимальной энергии системы при координации к катиону Ag+ двух I- анионов с противоположных сторон:
В дальнейшем ионная модель В. Косселя была дополнена (Фаянс, Ван-Аркель, Некрасов) поляризационными представлениями, учитывающих деформацию ионов и наличие постоянного или наведенного дипольного момента у нейтральных лигандов. С учетом поляризационных представлений ионная модель позволила качественно объяснить некоторые свойства комплексных соединений. В тоже время, многие свойства комплексов оставались не ясными – характеристические магнитные и оптические свойства, существование, наряду с тетраэдрическими комплексами, отвечающими минимальному отталкиванию 4-х лигандов, плоско-квадратных комплексов и т.д.
4.2. Метод валентных связей.
Согласно Дж. Льюису химическая связь осуществляется за счет общих электронных пар. В. Гейтлер и Ф. Лондон (1927 г.) на основе квантово-химического расчета молекулы Н2 обосновали предположение Дж. Льюиса и показали образование валентной химической связи обобщенной парой электронов с антипараллельными спинами. В дальнейшем Л. Полинг и Дж. Слейтер распространили эту модель, названную метод валентных связей (МВС) на многоатомные молекулы.
В рамках МВС химические связи М-L между комплексообразователем и лигандами:
1. Являются локализованными 2-х центровыми, 2-х электронными и образованы по донорно-акцепторному механизму.
2. Донорами электронной пары выступают лиганды, акцептором - комплексообразователь, предоставляя свободные валентные орбитали, гибридизованные в соответствии с пространственным положением лигандов вокруг комплексообразователя.
3. В зависимости от природы лигандов в гибридизации и образовании химической связи с лигандами могут участвовать внутренние (n-1)d или внешние nd валентные орбитали комплексообразователя, что приводит к существованию внутриорбитальных и внешнеорбитальных комплексов.
4. Участие внутренних (n-1)d валентных орбиталей металла в качестве свободных акцепторных орбиталей может приводить к изменению его числа неспаренных электронов и, как следствие этого, к различию магнитных свойств внутри- и внешнеорбитальных комплексов. Это явление называют магнитный критерий химической связи.
Рассмотрим описание химической связи в диамагнитном [Co(NH3)6]3+ и парамагнитном [CoF6]3- комплексах. В свободном состоянии Co(III) имеет 6 валентных электронов на внутренних валентных 3d орбиталях, 4 из которых неспаренны; валентные 4s, 4p и 4d орбитали Co(III) свободны. Парамагнетизм [CoF6]3- соответствует 4-м неспаренным электронам, что указывает на образование 6 донорно-акцепторных связей с фторидными лигандами, расположенными в вершинах октаэдра, в результате sp3d2 гибридизации внешних свободных 4s, 4p и 4d валентных орбиталей. Внутренние 3d орбитали Co(III) участия в образовании химических связей с лигандами не принимают и содержат 4 неспаренных электрона, что и приводит к парамагнетизму внешнеорбитального [CoF6]3- комплекса (рис. 4.1а).
| б |
| а |
Рис. 4.1. Электронное строение внешнеорбитального [CoF6]3- (а) и внутриорбитального [Co(NH3)6]3+ (б) комплексов.
Диамагнитный [Co(NH3)6]3+, являясь внутриорбитальным комплексом, не содержит неспаренных электронов на 3d орбиталях Co(III). В этом случае 6 донорно-акцепторных связей с аммиачными лигандами образуются в результате d2sp3 гибридизации двух 3d и 4s, 4p орбиталей Co(III), что приводит к спариванию всех 6-ти его электронов на трех не гибридизованных 3d орбиталях (рис. 4.1б).
Для комплексов с координационным числом 4 внешнеорбитальные и внутриорбитальные комплексы в результате sp3 и dsp2 гибридизации валентных орбиталей комплексообразователя отличаются не только магнитными свойствами но и тетраэдрической и плоско-квадратной структурой. Так, тетраэдрический [NiCl4]2- с sp3 гибридизацией 4s и 4р валентных орбиталей никеля является парамагнитным в результате наличия, подобно свободному иону Ni(II), 2-х неспаренных электронов на внутренних 3d орбиталях (рис. 4.2а).
Рис. 4.2. Электронное строение внешнеорбитального [NiCl4]2- (а) и внутриорбитального [Ni(CN)4]2- (б) комплексов.
В соответствии с dsp2 гибридизацией одной 3d, 4s и двух 4p валентных орбиталей никеля, плоско-квадратный комплекс [Ni(CN)4]2- не имеет неспаренных электронов на 3d орбиталя никеля и является диамагнитным.
В рамках МВС каждому пространственному расположению лигандов вокруг комплексообразователя соответствует свой тип гибридизации его валентных орбиталей (табл. 4.1). Таким образом, в МВС для обоснования специфики магнитных свойств и пространственного строения комплексов используется допущение о их внешне- и внутриорбитальном строении.
Табл. 4.1. Геометрическое строение комплексов и типы гибридизации валентных орбиталей комплексообразователя.
| Коорд. число | Гибридизация | Пространственное строение | Пример |
| sp | Линейное | [Cu(CN)2]- | |
| sp2 | Тригональное | [HgI3]- | |
| sp3 | Тетраэдр | [FeBr4]2- | |
| dsp2 | Плоский квадрат | [PtCl4]2- | |
| dsp3, d3sp | Тригональная бипирамида | [CuCl5]3- | |
| d2sp2, d4s | Квадратная бипирамида | [Ni(CN)5]3- | |
| d2sp3, sp3d2 | Октаэдр | [Co(CN)6]3- | |
| d3sp3, sp3d3 | Пентагональная бипирамида | [V(CN)7]4- | |
| d4sp3, sp3d4 | Квадратная антипризма | [TaF8]3- |
Несмотря на удовлетворительное объяснение магнитных свойств комплексов, квантовые расчеты электронного строения комплексов, выполненные в дальнейшем, показали, что допущение об участии внешних d орбиталей металла и образовании внешнеорбитальных комплексов требует больших энергетических затрат и вряд ли возможно. Основным недостатком МВС является использование представлений о локализованных двух центровых двух электронных связей металл-лиганд в комплексах, не применимых при рассмотрении химических связей с участием трех мерно делокализованных d орбиталей. В связи с этим, в настоящее время применение МВС для описания электронного строения комплексов носит достаточно характер.
4.3. Теория кристаллического поля.
Конценция изменения электронного строения ионов переходных металлов при действии электрического поля окружающих его заряженных частиц была предложена Беккерелем и в дальнейшем развита Х.А. Бете и Дж. Ван Флеком в начале XX в. К описанию электронного строения и свойств комплексных соединений эти представления были применены только в середине XX века Х. Хартманом и модель получила название «теория кристаллического поля» (ТКП).
Основные положения ТКП для комплексов переходных d металлов:
1. Комплекс существует и устойчив, благодаря электростатическому взаимодействию комплексообразователя с лигандами.
2. Лиганды рассматриваются без учета их электронного строения в качестве точечных зарядов или диполей.
3. Под действием электрического поля лигандов валентные пятикратно вырожденные (n-1)d орбитали расщепляются в зависимости от симметрии лигандного окружения.
4. Распределение валетных электронов металла по расщепленным (n-1)d орбиталям зависит от соотношения энергии спин-спаривания и энергии расщепления.
Рассмотрим, например, изменение энергии пятикратно вырожденных (n-1)d орбиталей центрального иона металла Мn+, находящегося в центре координат, под действием октаэдрического поля отрицательно заряженных лигандов [ML6]z, расположенных на осях координат (рис. 4.3). В результате отталкивания валентных электронов металла от отрицательно заряженных лигандов при равномерном распределении отрицательного заряда вокруг металла (сферически симметричное электрическое поле) энергия всех пяти d орбиталей повысится на величину Е0 по сравнению со свободным Мn+ ионом.
| Ион Мn+ в сферическом электрическом поле |
| Ион Мn+ в октаэдрическом электрическом поле лигандов |
| Свободный ион Мn+ |
Рис. 4.3. Расщепление валентных (n-1)d орбиталей металла в октадрическом поле лигандов.
Поскольку d орбитали имеют различную пространственную ориентацию, то при концентрации отрицательных зарядов на лигандах, расположенных на осях координат, повышение их энергии различается. Повышение энергии dz2 и dx2-y2 орбиталей, направленных к лигандам на осях координат, больше повышения энергии dxy, dxz и dyz орбиталей, направленных между осями координат.
Энергия расщепления D0 пятикратно вырожденных (n-1) орбиталей на двухкратно вырожденные dx2-y2,z2 орбитали и трехкратно вырожденные dxy,xz,yz орбитали называется параметром расщепления кристаллическим полем. Поскольку энергия расщепленных d орбиталей в октаэдрическом поле лигандов по сравнению со сферически симметричным электрическим полем не изменяется, то повышение энергии двух dx2-y2,z2 орбиталей происходит на 0.6D0 и понижение энергии трех dxy,xz,yz орбиталей на 0.4D0.
Для указания степени вырожденности и симметрии расщепленных под действием электрического поля лигандов орбиталей металла используют специальные символы. Трехкратно вырожденные и симметричные относительно центра симметрии и вращения вокруг осей координат dxy,xz,yz орбитали обозначают символом «t2g», тогда как двухкратно вырожденные и также симметричные относительно центра симметрии dx2-y2,z2 орбитали обозначают символом «eg». Таким образом, под действие октаэдрического электрического поля лигандов пятикратно вырожденные (n-1)d орбитали комплексообразователя расщепляются на различные по энергии трекратно и двухкратно вырожденные t2g и eg орбитали.
Рис. 4.4. Расщепление валентных (n-1)d орбиталей металла в тетраэдрическом поле лигандов.
Подобное рассмотрение изменения энергии пятикратно вырожденных (n-1)d орбиталей свободного иона металла при тетраэдрическом окружении лигандов в [ML4]z комплексах показывает (рис. 4.4) их расщепление также на двукратно (е) и трекратно (t) вырожденные орбитали, однако, с обратным энергетическим положением. Нижний индекс «g» при обозначении «е» и «t» орбиталей не указавается поскольку тетраэдрический комплекс не имеет центра симметрии. Уменьшение числа лигандов тетраэдрического комплекса по сравнению с октаэдрическим приводит к закономерному уменьшению параметра расщепления кристаллическим полем: DТ = 4/9DО.
Понижение симметрии лигандного окружения металла, например, тетрагональное искажение октаэдрических [ML6]z комплексов, связанное с удлинением металл-лиганд связей с аксиальными лигандами [ML4X2]z и образованием в предельном случае плоско-квадратных [ML4]z комплексов, приводит (рис. 4.5) к дополнительному расщеплению валентных (n-1)d орбиталей металла.
Рис. 4.5. Диаграмма расщепления (n-1)d орбиталей металла при тетрагональном искажении октаэдрических комплексов.
Заполнение валентными электронами расщепленных (n-1)d орбиталей металла происходит в соответствии с принципами Паули и минимума энергии. Для октаэдрических комплексов с d1, d2 и d3 электронной конфигурацией металла валентные электроны в соответствии с правилом Хунда заселяют t2g орбитали с параллльными спинами, приводя к t2g1, t2g2 и t2g3 электронной структуре комплексов.
Для металлов с d4 электронной конфигурацией три электрона также заселяют t2g орбитали с параллельными спинами. Заселение же четвертого электрона зависит от энергетических затрат на величину энергии спин спаривания (Есп.-сп.) при заселении t2g орбиталей с антипараллельным спином и нарушении правила Хунда, либо преодоления энергии расщепления кристаллическим полем Dо при заселении eg орбиталей с параллельным спином в соответствии с правилом Хунда. В первом случае образуется комплекс с t2g4 электронным строением и уменьшенным по сравнению со свободным металлом спиновой мультиплетностью 2S+1 = 3 (S – сумарный спин), называемых низкоспиновыми. При выполнении правила Хунда и заселении четвертого электрона на eg орбитали образуется комплекс с t2g3eg1 электронной структурой и подобной свободному металлу спиновой мультиплетностью 2S+1 = 5. Такие комплексы называют высокоспиновыми.
Аналогично, при распределении валентных d5, d6 и d7 электронов металлов по t2g и eg орбиталям октаждрических комплексов в зависимости от соотношения Есп.-сп. и Dо возможно образование двух типов комплексов:
- при Есп.-сп. > Dо обрауются высокоспиновые комплексы с электронной структурой металла t2g3eg2, t2g4eg2, t2g5eg2 в соответствии с правилом Хунда и спиновой мультиплетностью, подобной свободному металлу – 2S+1 = 6, 5, 4;
- Есп.-сп. < Dо образуются низкоспиновые комплексы с электронной структурой металла t2g5eg0, t2g6eg0, t2g6eg1 и пониженной по сравнению со свободным металлом спиновой мультиплетностью 2S+1 = 2, 1, 2.
Комплексы металлов с d8, d9 и d10 электронной конфигурацией характеризуются одним типом распределения электронов – t2g6eg2, t2g6eg3, t2g6eg4 со спиновой мультиплетностью, подобной свободному металлу: 2S+1 = 3, 2 и 0.
Таким образом, параметр D, характеризующий расщепление (n-1)d орбиталей металла под действием электрического поля лигандов, является одной из основных характеристик изменения свойств комплексов по сравнению со свободным ионом металла. Именно величина параметра D определяет для ряда электронных конфигураций металла определяет возможность образования высоко- или низкоспиновых комплексов с различным рапределением электронов по расщепленным орбиталям и различными свойствами.
Величина параметра расщепления кристаллическим полем D зависит от природы металла комплексообразователя, окружающих его лигандов и их пространственного положения вокруг комплексообразователя:
1. Лиганды в порядке увеличения параметра D для комплексов одного металла и подобного геометрического строения распологаются в так называемом спектрохимическом ряду: I- < Br- < Cl- < F- < OH- < C2O42- ~ H2O < NCS- < NH3 ~ En < NO2- < CN- < CO. В начале ряда расположены лиганды «слабого поля» - галогенид ионы, гидроксид и оксалат ионы, вода, образующие преимущественно высокоспиновые комплексы. Лиганды, в правой части ряда: окись углерода, цианид и нитрит ионы называются лигандами «сильного поля» и для них типично образование низкоспиновых комплексов. Для лигандов середины ряда – роданид иона, аммиака, этилендиамина в зависимости от природы металла образуются высоко- или низкоспиновые комплексы.
2. Увеличение эффективности действия электрического поля лигандов на d орбитали металла с увеличением их размера в ряду 3d << 4d < 5d, а также увеличения степени окисления металла приводит к увеличению параметра D в ряду: Mn(II) < Ni(II) < Co(II) < Fe(II) < V(II) < Fe(III) < Co(III) < Mn(IV) < Mo(III) < Rh(III) < Ru(III) < Pd(IV) < Ir(III) < Pt(IV).
3. Праметр D для тетраэдрических комплексов составляет только 4/9 от параметра D октаэдрических комплексов.
Комплексы «тяжелых» 4d и 5d металлов практически не зависимо от природы лигандов образуют преимущественно низкоспиновые комплексы, тогда как образование низко- или выскоспиновых комплексов «легких» 3d металлов в основном определяется силой поля лигандов.
В отличие от МВС, теория кристаллического поля для обоснования различия магнитных свойств комплексов одного и того же иона металла с различным лигандным окружением, например, диамагнитного [Fe(CN)6]4- и парамагнитного [Fe(H2O)6]2+ не использует гипотезу о их внутриорбитальном (d2sp3 гибридизация) и требующем больших энергетических затрат внешнеорбитальном (sp3d2 гибридизация) строении. Различие в магнитных свойствах определяется низко- и высокоспиновым характером распределения 6-ти валетных электронов Fe(II) по расщепленным t2g и eg орбиталям (рис. 4.6). Являясь лигандами сильного и слабого поля, цианид-ионы и молекулы воды образуют с Fe(II) низко- и высокоспиновые комплексы с t2g6eg0 и t2g4eg2 распределением электронов, что и определяет диамагнетизм [Fe(CN)6]4- и парамагнитизм [Fe(H2O)6]2+ комплексов.
Рис. 4.6. Диамагнетизм [Fe(CN)6]4- и парамагнитизм [Fe(H2O)6]2+ комплексов.
Расщепление пятикратно вырожденных (n-1)d орбиталей металла в комплексах и изменение параметра D в зависимости от природы лигандов определяет характерную окраску комплексов как в твердом состоянии, так и в растворах. При поглощении комплексом электромагнитного излучения в видимой области спектра (400-750) нм, энергия квантов которого Е = hn = hc/l равна величине D, происходит перенос электрона с t2g на eg орбитали. Не поглощенное электромагнитное излучение видимой области спектра и определяет окраску комплекса в соответствии с «цветовым кругом Ньютона» (рис. 4.7), показывающим основной и дополнительный цвет видимого излучения.
| Окраска |
| Фиолетовый |
| Поглощение |
| Желтый |
| 490 нм |
| Голубой |
| 430 нм |
| Зеленый |
| Красный |
| 400 нм |
| 800 нм |
| 560 нм |
| 600 нм |
| 650 нм |
| Оранжевый |
Рис. 4.7. Цветовой круг Ньютона.
Например, аквакомплекс титана(III) [Ti(H2O)6]3+ c t2g1eg0 электронным распределением в результате фотовозбуждения, соответствующего переходу электрона на более высокоэнергетические eg орбитали:
[Ti(H2O)6]3+ (t2g1eg0) + hn = *[Ti(H2O)6]3+ (t2g0eg1)
поглощает кванты света в желтой области спектра, что приводит к его фиолетовой окраске. Изменение лигандного окружения иона металла в соответствии с положением лиганда в спектрохимическом ряду приводит к изменению параметра D и, как следствие этого, к изменению энергии и длины волны поглощаемых комплексом квантов и к характеристической окраске комплекса – например, в ряду [CuCl4]2-, [Cu(H2O)4]2+, [Cu(NH3)4]2+ цвет комплексов изменяется от зеленого к голубому и фиолетовому.
Наряду с энергией расщепления кристаллического поля D, важную роль в ТКП играет также энергия стабилиции кристаллическим полем (ЭСКП)– выйгрыш в энергии при распределении электронов по расщепленным в комплексе (n-1)d орбиталям металла по сравнению с энергией пятикратно вырожденных (n-1)d орбиталей металла в эквивалентном сферическом электрическом поле.
Табл. 4.2. ЭСКП октадрических и тетраэдрических комплексов.
| Mn+ | Октаэдрические комлексы | Тетраэдрические комплексы | |
| Низкоспиновые | Высокоспиновые | Высокоспиновые | |
| d1 | 0.4Do | 0.6Dт | |
| d2 | 0.8Do | 1.2Dт | |
| d3 | 1.2Do | 0.8Dт | |
| d4 | 1.6Do | 0.6Do | 0.4Dт |
| d5 | 2.0Do | 0Do | 0Dт |
| d6 | 2.4Do | 0.4Do | 0.6Dт |
| d7 | 1.8Do | 0.8Do | 1.2Dт |
| d8 | 1.2Do | 0.8Dт | |
| d9 | 0.6Do | 0.4Dт | |
| d10 | 0Do |
Оценку величины ЭКСП комплекса получают на основании диаграмм расщепления (n-1)d орбиталей металла (рис. 4.3, 4.4) в электрическом поле лигандов, показывающих уменьшение или повышение энергии системы по сравнению со сферическим электрическим полем при заселении электронами расщепленных (n-1)d орбиталей. Для октаэдрических [ML6]z комплексов (рис. 4.3) заселение каждым электроном t2g орбиталей приводит к выйгрышу энергии системы на 0.4Dо, заселение же eg требует затрат энергии 0.6Dо. Для тетраэдрических [ML4]z комплексов с противоложным энергетическим положением e и t орбиталей металла (рис. 4.4) заселение каждым электроном расщепленных e и t орбиталей сопровождается понижением и повышением энергии системы на 0.6Dт и 0.4Dт.
Являясь отражением термодинамической устойчивости комплеков, оценки их величины ЭСКП (табл. 4.2) согласуются с экспериментальными данными изменения энергии кристаллической решетки для высокоспиновых гексафторидных комплексов 3d металлов (рис. 4.8).
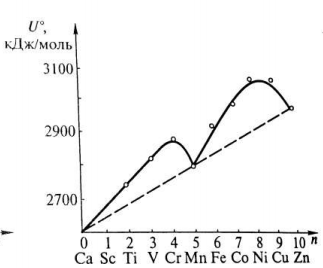
Рис. 4.8. Изменение энергии кристаллической решетки гексафторидных комплексов 3d металлов.
Величины ЭСКП позволяют установить наиболее предпочтительный координационный изомер, например [Cu(NH3)6][NiCl4] или [Ni(NH3)6][CuCl4]. Для этого рассчитывают разницу ЭСКП для комплексного катиона и аниона изомеров. Величина ЭСКП [Cu(NH3)6]2+ и [NiCl4]2- составляет 0.6Dо и 0.8Dт соответственно. Учитывая, что Dт = 4/9Do, разница между величинами ЭСКП [Cu(NH3)6]2+ и [NiCl4]2- будет составлять 19/45Do. Аналогично, величины ЭСКП [Ni(NH3)6]2+ и [CuCl4]2- составляет 1.2Dо и 0.4Dт, а разница между ними 28/45Do. Большая разница ЭСКП комплексных катиона [Ni(NH3)6]2+ и аниона [CuCl4]2- по сравнению с [Cu(NH3)6]2+ и [NiCl4]2- показывает более предпочтительное образование изомера состава [Ni(NH3)6][CuCl4].
Наряду с магнитными и оптическими свойствами, влияния электронного строения металла на термодинамическую устойчиость комплексов, ТКП предсказывает искажение геометрического строения комплексов при не равномерном распределении электронов по расщепленным (n-1)d орбиталям металла. В отличие от правильного октаэдрического строения [Co(CN)6]3- с t2g6eg0 электронным распределением, тетрагональное искажение аналогичного комплекса [Cu(CN)6]4- с t2g6eg3 электронным распределением, содержащего 3 электрона на 2-х кратно вырожденных eg орбиталях, приводит к эффективной трансформации октаэдрического в плоско-квадратный комплекс:
[Cu(CN)6]4- ® [Cu(CN)4]2- + 2CN-.
Все выше сказанное показывает, что относительная простота и широкие возможности ТКП для объяснения и прогнозирования физико-химических свойств комплексов определяют большую популярность это модели описания химической связи в комплесных соединениях. В тоже время, акцентируя внимание на изменении электронной структуры металла при комплексообразовании, ТКП не учитывает электронное строение лигандов, рассматривая их в качестве точечных отрицательных зарядов или диполей. Это приводит к ряду ограничений ТКП при описании электронного строения комплексов. Например, в рамках ТКП трудно объяснить положение ряда лигандов и металлов в спектрохимических рядах, что связано с определенной степенью ковалентности и возможность образования кратных металл-лиганд связей. Эти ограничения устраняются при рассмотрении электронного строения комплексных соединений более сложным и менее наглядным методом молекулярных орбиталей.
4.4. Метод молекулярных орбиталей. Теория поля лигандов.
Применение метода молекулярных орбиталей (ММО) к описанию электронного строения комплексных соединений было развито Оргелом, Йоргенсеном и Бальхаузеном в середине 50-х годов XX в. и получило название «теория поля лигандов» (ТПЛ). ТПЛ описывает образование комплекса и снятие вырождения d орбиталей металла не только за счет электростатического взаимодействия с лигандами, но и за счет перекрывания орбиталей металла и лигандов (определенной степени ковалентного связывания) с образованием делокализованных молекулярных орбиталей.
При построении молекулярных орбиталей комплекса используют традиционные приближения:
1. Адиабатическое – комплекс рассаматривается при фиксированных координатах металла и лигандов без учета их колебаний.
2. Валентное – в образовании молекулярных орбиталей принимают участие только валентные орбитали металла и групповые валентные орбитали лигандов.
3. Построение молекулярных орбиталей комплекса проводят, используя линейную комбинацию валентных орбиталей металла и групповых орбиталей лигандов.
Рассмотрим электронное строение октаэдрических комплексов с s связями металл-лиганд – например, [M(NH3)6]z+. Валентными орбиталями иона металла Mz+ являются (n-1)d-, ns- и np-орбитали, содержащие q электронов (q = 1 – 9). Каждая из 6 молекул NH3, выступающих в качестве s-донорных лигандов, характеризуется наличием электронной пары на sp3-гибридной орбитали донорного атома азота. Таким образом, в образовании МО комплекса участвуют 15 [9(Mz+) + 6(NH3)] исходных орбиталей, содержащих (q+12) электронов.
Октаэдрическое строение комплекса определяет расположение лигандов на осях X, Y, Z системы координат, характеризующихся 6-ю эквивалентными групповыми орбиталями, наиболее эффективно перекрывающихся с валентными орбиталями иона металла.
Перекрывание сферической ns орбитали металла с групповой орбиталью лигандов, состоящей и суммы всех 6-ти sp3 гибридных орбиталей лигандов, приводит (рис. 4.9) к образования ss связывающей и ss* разрыхляющей МО.
Рис. 4.9. Образование ss связывающей и ss* разрыхляющей МО.
Ориентированные по осям координат nрx,y,z орбитали металла перекрываются с орбиталями лигандов на осях X, Y и Z (рис. 4.10) с образованием 3-х кратно вырожденных связывающих и разрыхляющих МО sx,y,z и s*x,y,z.

Рис. 4.9. Образование sy связывающей и sy* разрыхляющей МО.
Перекрывание dx2-y2 и dz2 орбиталей металла с групповыми орбиталями лигандов (рис. 4.10) приводит к образованию 2-х кратно вырожденных sz2,x2-y2 связывающих и s*z2,x2-y2 разрыхляющих МО.
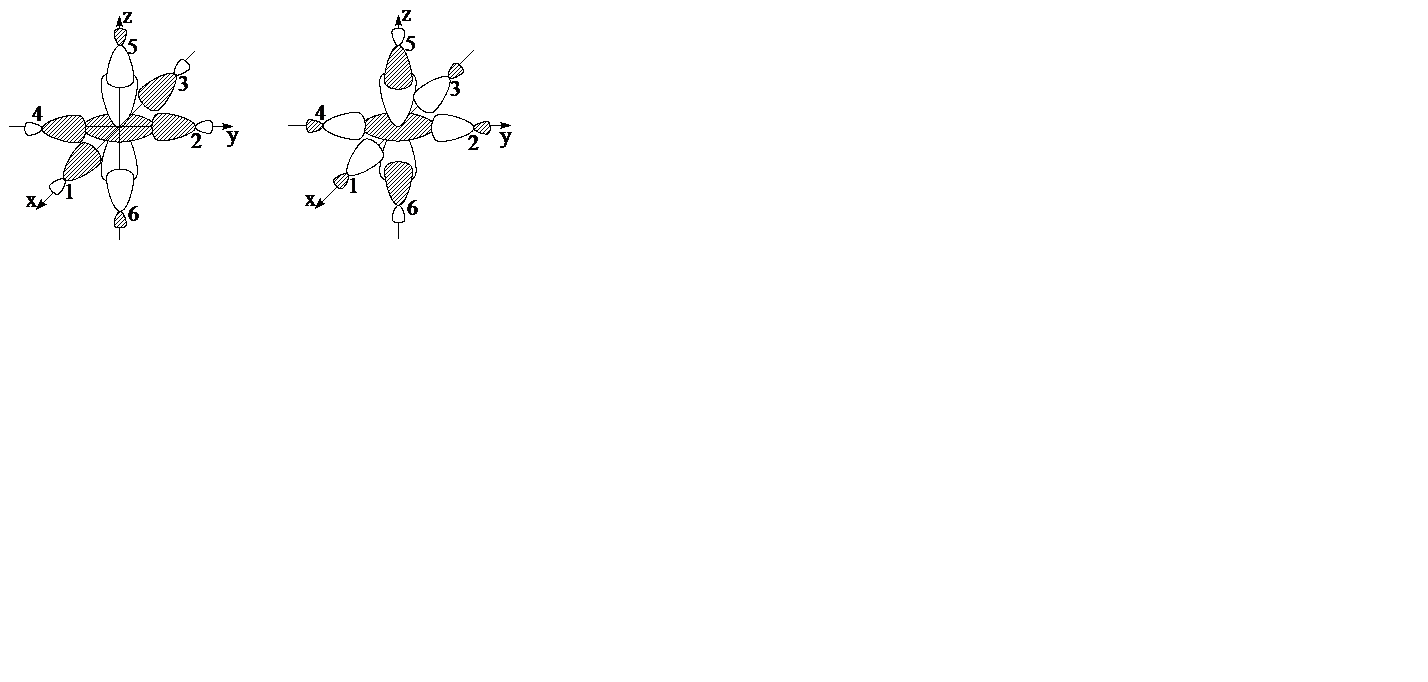
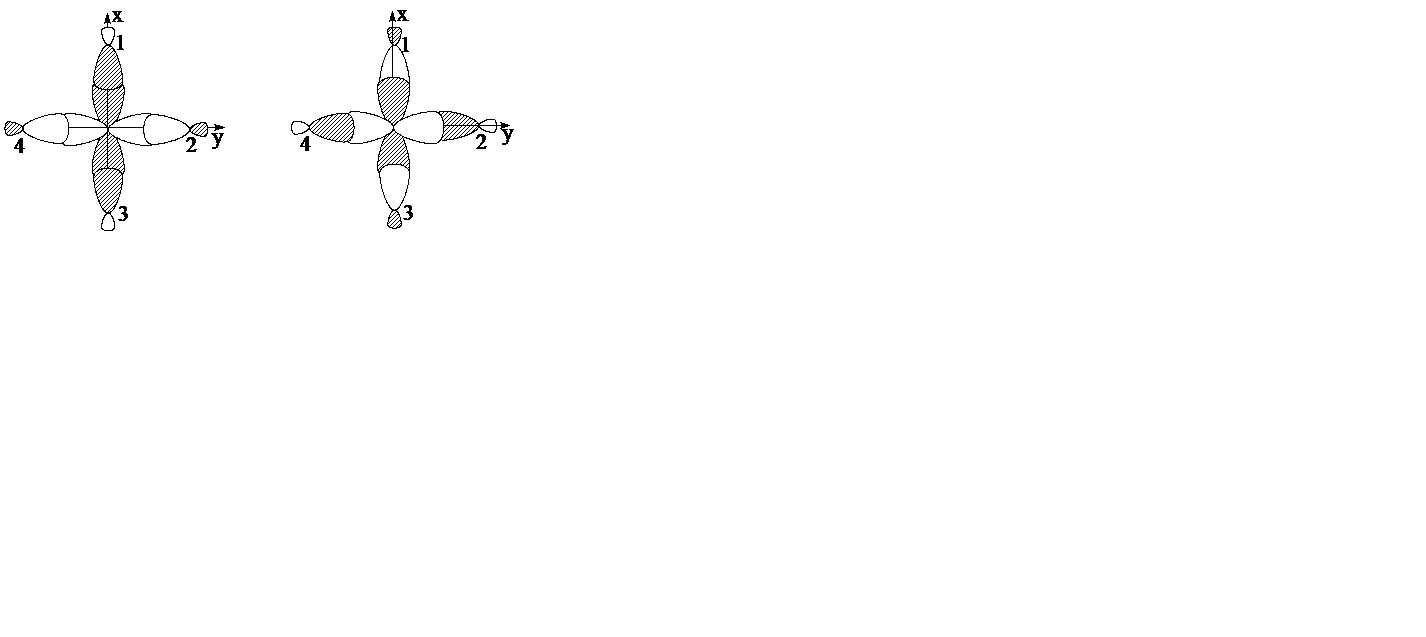
Рис. 4.10. Образование sz2,x2-y2 связывающей и s*z2,x2-y2 разрыхляющей MO.
Орбитали металла dxy, dzy, dzx, не перекрывающиеся с орбиталями лигандов (рис. 4.11), образуют 3-х кратно вырожденные несвязывающие nxy,zy,zx МО.

Рис. 4.11. Образование несвязывающей nxy,zy,zx МО.
Таким образом, электронное строение [M(NH3)6]z+ описывается 12-ю делокализованных 7-центровых МО и 3-х несвязывающих МО, локализованных на металле (рис. 4.12). Различие в энергетическом положении валентных орбиталей иона металла и групповых орбиталей лигандов определяет различный вклад их волновых функций в МО комплекса – преимущественную локализацию s-связывающих МО на лигандах и s*-разрыхляющих на металле.
| Групповые орбитали лигандов |
Рис. 4.12. Качественная диаграмма МО [M(NH3)6]z+ комплексов.
Распределение (12+q) электронов по МО комплекса в соответcтвии с принципом Паули и минимума энергии определяет электронную формулу [M(NH3)6]3+: (ss)2(sx,y,z)6(sx2-y2,z2)4(nxy,xz,yz)x(s*x2-y2,z2)y (x+y=q). Таким образом, независимо от природы иона металла электронное строение [M(NH3)6]3+ комплексов характеризуется наличием 12 электронов на s связывающих орбиталях, преимущественно локализованных на лигандах. Заполнение несвязывающих nxy,xz,yz (t2g) и разрыхляющих s*x2-y2,z2 (e*g) орбиталей, преимущественно локализованных на металле, зависит от числа электронов металла и энергетического зазора D между t2g и e*g орбиталями.
Для ионов металлов с d1, d2, d3 электронной конфигурацией (q = 1, 2, 3) минимальной энергии комплекса отвечает последовательное заполнению электронами t2g орбиталей в соответствии с правилом Хунда – (t2g)1, (t2g)2, (t2g)3. При q = 4, 5, 6, 7 в зависимости от соотношения энергетического зазора D и энергии спин-спаривания (Есп.-сп.) минимуму энергии системы отвечают две различные электронные конфигурации, соответствующие низко- и высокоспиновым комплексам.
При D > Eсп.-сп. заполняются электронами t2g орбитали – (t2g)4, (t2g)5, (t2g)6 и после этого разрыхляющие e*g орбитали, приводя при q = 7 к электронной конфигурации (t2g)6(e*g)1. При D < Eсп.-сп., для q = 4, 5 в соответствии с правилом Хунда происходит последовательное заполнение электронами разрыхляющих e*g орбиталей – (t2g)3(e*g)1(t2g)3(e*g)2 и только для q = 6, 7 дальнейшее заполнение t2g орбиталей – (t2g)4(e*g)2, (t2g)5(e*g)2. Для q = 8, 9, 10 минимальной энергии комплекса соответствуют электронные конфигурации (t2g)6(e*g)2, (t2g)6(e*g)3 и (t2g)6(e*g)4.
| МВС |
| ТКП |
Рис. 4.13. Электронное строение [M(NH3)6]z+ в рамках ТПЛ, ТКП, МВС.
Сравнение описания электронного строения октаэдрических комплексов в рамках методов ТПЛ, ВС и ТКП показывает (рис. 4.13), что метод МО дает наиболее общий подход, включая методы ВС и ТКП как частные случаи. Шести электронным парам лигандов на связывающих ss, sx,y,z и sx2-y2,z2 МО в рамках метода ВС отвечает шесть донорно-акцепторных s-связей с участием d2sp3 гибридных орбиталей металла. Несвязывающие nxy,xz,yz и разрыхляющие s*x2-y2,z2 МО соответствуют расщепленным в октаэдрическом поле лигандов dxy,xz,yz (t2g) и dx2-y2,z2 (eg) орбиталям металла.
Качественное согласие характера расщепления (n-1)d орбиталей металла на t2g и eg орбитали, разделенных энергетическим зазором D и распределения электронов между ними, приводящим к существованию низкоспиновых и высокоспиновых коплексов показывает, что в рамках ТПЛ могут быть использованы параметры ТКП – параметр расщепления кристаллическим полем, энергия стабилизации кристаллическим полем, спектрохимический ряд лигандов.
Сохраняя все достоинства ТКП, теория поля лигандов позволяет получить более строгое и полное описание электронного строения комплексов. ТПЛ показывает, что характер химической связи в комплексных соединениях связан не только с кулоновским взаимодействием между ионом металла и лигандами, но и с определенной ковалентной составляющей. Это приводит к перераспределению электронной плотности между металлом и лигандами в результате образования связывающих и разрыхляющих МО. Расщепленные в рамках ТКП (n-1)d орбитали металла, в ТПЛ являются молекулярными орбиталями, имеющими смешанный металл-лигандный характер. Для [ML6]z комплексов с s связями металл-лиганд, несмотря на преимущественно металлический характер, eg орбитали являются s* разрыхляющими, а t2g – несвязывающими МО, способными участвовать в p связях с лигандами. Между лигандами и ионом металла, наряду с s донорно-акцепторным ML взаимодействием, возможны дополнительное p донорное и p-акцепторное взаимодействия лигандов с металлом с участием dxy,xz,yz орбиталей.
В комплексах с s и p связями металл-лиганд к МО s- и s*-типа добавляются МО p- и p*-типа. Лиганды p-доноры характеризуются наличием низкоэнергетических заполненных электронами орбиталей p-типа, которые при взаимодействии dxy,xz,yz орбиталями образуют трехкратно вырожденные t2g связывающие и t*2g разрыхляющие МО, в основном локализованные на лигандах и меенн. Наоборот, p-акцепторные лиганды, характеризуются наличием свободных орбиталей p-типа, взаимодействие которых с dxy,xz,yz орбиталями металла приводит к образованию t2g связывающих МО, преимущественно локализованных на металле, и разрыхляющих t*2g МО, в основном локализованных на лигандах (рис. 4.14).
| L - s и p донор |
| L - s донор |
| L - s донор, p акцептор |
Рис. 4.14. Влияние металл-лиганд p связей на величину параметра D [ML6]z комплексов.
ТПЛ показывает, что влияние лигандов на величину D и их положение в спектрохимическом ряду определяется как s, так и p свойствами лигандов - s-донорные свойства лигандов определяют положение e*g орбиталей, тогда как p-донорно-акцепторные свойства – положение t2g орбиталей. Для лигандов с s-донорными свойствами dxy,xz,yz орбитали металла в комплексе являются несвязывающими t2g МО, тогда как p донорные и p акцепторные лиганды, характеризующиеся заполненными и свободными орбиталями p-типа, эффективно взаимодействуют с dxy,xz,yz орбиталями металла. Это приводит к трансформации несвязывающих t2g орбиталей металла либо в разрыхляющие более высоко лежащие t*2g орбитали для p-донорных лигандов, либо в связывающие более низко лежащие t2g орбитали для p-акцепторных лигандов. Именно совместный характер s- и p-взаимодействия лигандов с металлом определяет их положение в спектрохимическом ряду: I- < Br- < Cl- < F- < OH- < C2O42- ~ H2O < NCS- < NH3 ~ En < NO2- < CN- < CO
| s доноры, p доноры |
| s доноры, p акцепторы |
| s доноры |
В качестве комплексов с s- и p-связями рассмотрим электронное строение [CoF6]3- и [Co(CN)6]3-. Образование МО [CoF6]3- происходит в результате взаимодействия 9 валентных орбиталей Co(III) 3d64s04p0 и 18 валентных 2р6 орбиталей ионов F-. Значительное энергетическое различие валентных орбиталей Co(III) и 2s орбиталей F- позволяет пренебречь взамодействием. Таким образом, электронное строение [CoF6]3- характеризуется наличием 27 МО, на которых распределены 42 электрона. Наряду с образованием ss, sx,y,z, sx2-y2,z2 связывающих и s*s, s*x,y,z, s*x2-y2,z2 разрыхляющих МО, перекрывание dxy, dxz, dyz орбиталей Co(III) c р орбиталями F-, перпедикулярных осям координат (рис. 4.15),
Рис. 4.15. Образование p связывающих и p* разрыхляющих МО [CoF6]3-.
Образуются 15 МО – 2 делокализованных трехкратно вырожденных pxy,xz,yz связывающих и p*xy,xz,yz разрыхляющих и 9-ти кратно вырожденные несвязывающие np. Малая величина энергетического различия между p*xy,xz,yz и s*x2-y2,z2 орбиталями (малая величина D) приводит к распределению 42 электронов по МО комплекса в соответствии с правилом Хунда и высокоспиновому характеру [CoF6]3- (рис. 4.16).
Рис. 4.16. Качественная диаграмма МО [CoF6]3- и [Co(СN6]3-.
В отличие от F- лигандов с s- и p-донорными свойствами, CN- ионы - s донорные и p-акцепторные лиганды. Это связано с наличием у CN- иона свободных p* орбиталей, способных к перекрыванию с dxy,xz,yz орбиталями Co(III) и образованию 9 МО: трехкратно вырожденных pxy,xz,yz связывающих, p*xy,xz,yz разрыхляющих и несвязывающих np. Дативное металл®лиганд p-взаимодействие приводит к увеличению параметра D, что определяет низкоспиновый характер распределения электронов [Co(СN6]3- (рис. 4.16).
Наряду с октаэдрическими, для 3d меенны с p донорными лигандами слабого поля характерно образование тетраэдрических комплексов. В тетраэдрических TiCl4, VCl4, [CrO4]2-, [MnO4]- в образовании химических связей принимают участие 21 атомная орбиталь: 3d-, 4s- и 4p-орбитали иона металла, содержащие q электронов, и 12 АО лигандов (по 3 р-орбитали от каждого из лигандов), содержащие 24 электрона. Образование ss, s*s и трехкратно вырожденных sx,y,z, s*x,y,z происходит в результате перекрывания 4s- и 4p-орбиталей металла с групповыми орбиталями лигандов. Взаимодействие 3dxy,xz,yz, 3dx2-y2,z2 орбиталей металла с групповыми орбиталями лигандов определяет образование трех- и двухкратно вырожденных pxy,xz,yz, p*xy,xz,yz и px2-y2,z2, p*x2-y2,z2 МО. Оставшиеся групповые орбитали лигандов образуют трехкратно вырожденные несвязывающие np МО (рис. 4.17). Распределение (24+q) электронов по 21 МО приводит к общей электронной формуле: (ss)2(sx,y,z)6(pxy,xz,yz)6(px2-y2,z2)4(np)6(p*x2-y2,z2)x(p*xy,xzy,yz)y(s*s)0(sx,y,z)0 (x + y = q).
| Групповые орбитали лигандов |
Рис. 4.17. Качественная диаграмма МО тетраэдрических комплексов.
Таким образом, в тетраэдрических комплексах независимо от природы иона металла 24 электрона лигандов заселяют s/p-связывающие и несвязывающие МО в основном локализованных на лигандах. Валентные d электроны металла заполняют более высоколежащие разрыхляющие p*x2-y2,z2 и p*xy,xzy,yz МО. В отличие от ТКП, связывающей окраску комплексов с оптическими переходами между расщепленными e и t d орбиталями металла, ТПЛ учитывает также оптические переходы, называемыми переходами переноса заряда лиганд-металл. Так, фиолетовая и оранжевая окраска [MnO4]- и [CrO4]2-, содержащих металлы в высшей степени окисления и характеризующихся свободными p*x2-y2,z2 € и p*xy,xzy,yz (t) МО отсутствие обусловлена оптическим переходом переноса заряда между заполненными np и свободными p*x2-y2,z2 МО.
| Групповые орбитали лигандов |
Рис 4.18. Качественная диаграмма МО плоско-квадратных комплексов.
Для комплексов 3d металлов с лигандами сильного поля и 4d, 5d металлов независимо от природы лигандов образуются плоско-квадратные комплексы. Перекрывание валентных (n-1)dx2-y2, (n-1)dz2, ns, npx и npy орбиталей металла с групповыми орбиталями лигандов на осях Х и У приводит к образованию связывающих ss, sx,y, sx2y2, sz2 и разрыхляющих ss*, sx,y*, sx2y2*, sz2* МО (рис. 4.18). Орбитали (n-1)dxy, (n-1)dxz, (n-1)dyz участвуют в образовании p-МО. Причем, (n-1)dxy,-орбиталь перекрывается с 4-мя p орбиталями лигандов, тогда как эквивалентные (n-1)dxz и (n-1)dyz орбитали только с 2-мя. Валентная nрz орбиталь металла и 4 групповые орбитали лигандов образуют несвязывающие МО.
Например, для [PtCl4]2- распределение по МО 32 валентных электрона (24 лигандов и 8 меенн) приводит к электронной формуле [PtCl4]2-: (ss)2(sx,y)4 (sx2y2)2(sz2)2(pxy)2(pxz,yz)4(np)8(p*xz,yz)4(s*z2)2(p*xy)2(s*x2-y2)0(npz)0(s*s)0(sx,y)0. Комплекс диамагнитен и в соответствии электронами на связывающих и разрыхляющих МО характеризуется единичной кратностью индивидуальной связи Pt-Cl (К = (16-8)/(2×4) = 1).
Рассмотрение моделей химической связи комплексов показывает, что, несмотря на простоту и наглядность МВС и ТКП, более сложная и менее наглядная модель ТПЛ, рассматривающая делокализованные лиганд-металл химические связи в результате образования молекулярных орбиталей, безусловно является наиболее строгой и полной моделью, описывающей физико-химические свойства комплексов.
Контрольные вопросы.
1. В рамках ионной модели обоснуйте линейное, плоско-треугольное, тетраэдрическое и октаэдрическое строение комплексов с координационным числом 2, 3, 4 и 6.
2. Какой тип гибридизации валентных орбиталей комплексообразователя соответствует образованию комплексов следующей структуры: а) линейной, б) плоско-треугольной, в) тетраэдрической, в) плоско-квадратной, г) тригонально-бипирамидальной, д) квадратно-бипирамидальной, е) октаэдрической?
3. В рамках метода ВС опишите электронное строение, определите геометрическое строение, магнитные свойства и мультиплетность следующих комплексов: а) [CuCl2]-, б) [NiCl4]2-, в) [Ni(CN)4]2-, г) [PdCl4]2-, д) [Fe(H2O)6]2+, е) [Fe(CN)6]4-, ж) [IrCl6]3-.
4. Приведите примеры лигандов: s доноров, s и p доноров, s доноров и p акцепторов. Для каких из этих лигандов характерно образование дативной лиган-металл химической связи?
5. В чем причина расщепления пятикратно вырожденных d орбиталей комплексообразователя на более высокоэнергетические dx2-y2,z2 и более низкоэнергетические dxy,xz,yz орбитали?
6. Что называется параметром расщепления кристаллическим полем? От каких параметров комплекса он зависит?
7. Что такое спектрохимический ряд лигандов? С чем связано его название?
8. Какой комплекс имеет большее значение D: а) [Co(CN)6]3- или [Co(NH3)6]3+, б) [Co(H2O)6]3+ или [Rh(H2O)6]3+, в) [PtI4]2- или [PtCl4]2-, г) [NiCl4]2- или [PdCl4]2-?
9. Определите какие из комплексов: [CuCl4]2-, [Cu(CN)4]2-, [Ni(CO)4], [FeCl4]-, [RhCl4]3-, [RuCl6]3- являются низкоспиновыми и какие высокоспиновыми?
10. Обоснуйте в рамках ТКП изменение характерной окраски следующих комплексов: [CuCl2]- - бесцветный, [Cu(H2O)4]2+ - голубой, [Cu(CN)4]2- - бесцветный.
11. Что такое энергия стабилизации кристаллическим полем? Рассчитайте величину ЭСКП [Co(CN)6]3-, [Co(CN)6]4-, [Co(H2O)6]3+, [Co(H2O)6]2+ комплексов и сделайте вывод об окислительно-восстановительных свойствах Co(III) и Co(II) в цианидном и аква- окружении.
12. Обоснуйте следующие экспериментальные данные: а) комплексы Cr(III) независимо от лигандного окружения всегда парамагнитны, б) комплексы Zn(II) всегда диамагнитны, в) комплекс [Co(NO2)6]3- диамагнитен, тогда как комплекс [CoF6]3- парамагнитен.
13. В чем отличие t2g и eg орбиталей при рассмотрении электронного строения октаэдрического комплекса в рамках ТКП и ТПЛ?
14. Почему, несмотря на меньшее значение дипольного момента СО (m = 0.10 D) по сравнению с H2O (m= 1.83 D), CO – лиганд сильного поля, а H2O – лиганд слабого поля?
15. В рамках ТПЛ опишите электронное строение и определите кратность связи метал-лиганд для оксокомплексов марганца: [MnO4]-, [MnO4]2-, [MnO4]3-. Сделайте вывод о характере изменения устойчивости в ряду оксокомплексов Mn(VII), Mn(VI) и Mn(V)?
16. Определите природу оптических полос поглощения, определяющих окраску комплексов: [V(H2O)6]3+ - зеенный, [VF6]- - бесцветный, [VO4]3- - светло-желтый.
17. Сопоставьте в рамках метода ВС, ТКП и ТПЛ описание электронного строения, кратность связи метал-лиганд, магнитные и оптические свойства следующих комплексов: [Cr(NH3)6]3+, [Cr(CN)6]3-, [CrO4]2-. Какая из моделей и почему дает более адекватное описание электронного строения и свойств комплексов?
Дата добавления: 2016-01-03; просмотров: 4142;