Соли и комплексные соединения элементов 9 группы
Простые неорганические соединения наиболее устойчивы у кобальта в степени окисления +2, у родия +3 и у иридия +4. Таким образом, здесь наблюдается общая тенденция увеличения наиболее стабильной степени окисления при движении вниз по группе. Стандартный электродный потенциал пары Co(III)/Co(II) сильно зависит от природы лиганда:
E°, В
[Co(H2O)6]3+ + e– ® [Co(H2O)6]2+ +1.95
[Co(NH3)6]3+ + e– ® [Co(NH3)6]2+ +0.1
[Co(CN)6]3– + e– ® [Co(CN)6]4– –0.83
Стабилизация кристаллическим полем делает более благоприятным образованием кобальта(III) и ослабляет его окислительные свойства. Таким образом, у кобальта в степени окисления + 3 очень богатая химия координационных соединений, в то время как его простые неорганические соли неустойчивы в водных растворах, так как являются сильными окилсителями. Акцепторные лиганды стабилизируют все три металла в низших степенях окисления. Особенно разнообразна химия родия(I) и иридия(I).
ДОПОЛНЕНИЕ. Активация С-Н связей.
Связи С-Н отличаются низкой полярностью, обладают высокой прочностью (Е = 470 кДж/моль), что приводит к низкой реакционной способности углеводородов. Важнейшей задачей нефтехимии является перевод углеводородного сырья в практически важные соединения, содержащие кратные связи или функциональные группы. Гомогенным катализом с участием комплексов переходных металлов эти процессы удается осуществлять с хорошим выходом и высокой селективностью в мягких условиях. В основе действия металлокомплексного катализатора лежит процесс активации С-Н связи. Молекула катализатора, благодаря возникновению взаимодействия М...С-Н, где М – атом металла, входящего в состав катализатора, а С-Н – фрагмент углеводорода, делает связи С-Н и С-С более реакционноспособными. Это и называют активацией связи (А.Е. Шилов, Г.Б. Шульпин, Активация и каталитические реакции углеводородов, М., Наука, 1995). Активация органической молекулы повышает ее реакционную способность в процессах замещения и присоединения. Внедрение молекул СО по связи металл-углерод приводит к формированию карбоксильной группы:

Реакции такого типа лежат в основе процессов гидроформилирования:

В некоторых случаях в результате С-Н активации проявляется циклометаллирование – внутримолекулярное окислительное присоединение или реакция замещения, сопровождающаяся образованием связи металл-углерод:

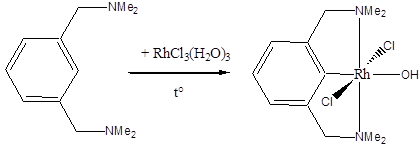
(A. van der ZeijdenG., van Koten et al, Inorg. Chem., 1988, 27, 1014). Как правило, оно осуществляется в орто-положение бензольного ядра. Как видно из последнего примера, ортометаллирование может протекать и в комплексах металлов в сравнительно высоких степенях окисления, например, родия(III) и платины(IV).
КОНЕЦ ДОПОЛНЕНИЯ
Соединения в степени окисления –1, 0, +1.
Химия этих элементов представлена преимущественно комплексами с π-акцепторными лигандами, устойчивость которых возрастает вниз по группе по мере увеличения размеров d-орбиталей. Общий метод синтеза основан на восстановлении соединений кобальта(II), родия(III) и иридия(IV):
CoCl2 + 4PMe3 + 2Na = Co(PMe3)4 + 2NaCl.
2RhCl3×3H2O + 6CO = Rh2(CO)4Cl2 + 2COCl2 + 6H2O.
Соединения родия(I) и иридия(I) с фосфинами, алкенами, циклоалкадиенами в сухом виде устойчивы к окислению кислородом воздуха, хорошо растворимы в большинстве органических растворителей. Они имеют плоско-квадратную геометрию, соответствующую их электронной конфигурации d8, которая подробно обсуждается ниже на примере соединений никеля(II). Характерным типом реакций является окислительное присоединение, осуществимое при их гидрировании, гидрохлорировании, а иногда просто при растворении в хлор-содержащих растворителях, например, хлороформе или дихлорметане:
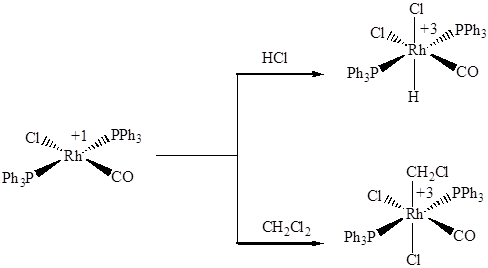
Благодаря легкости протекания обратной реакции – восстановительного элиминирования, многие из этих соединений активно катализируют гидрирование и гидроформилирование олефинов. Для этих целей часто используют трис(трифенилфосфин)родий(I) хлорид Rh(PPh3)3Cl, называемый катализатором Уилкинсона, и бис(трифенилфосфин)карбонилродий(I) хлорид транс-Ir(PPh3)2COCl – катализатор Васка. Смешанные иодидо-карбонильные комплексы родия, образующиеся при взаимодействии трихлорида родия с СО и иодоводородом в метаноле, являются интермедиатами в процессе производства уксусной кислоты, разработанном американской компанией Monsanto Industry в 1970-е годы:
RhCl3, I–
CH3OH + CO ¾¾¾¾¾® CH3COOH
180 °C, 40 атм
В настоящее время по этой технологии, пришедшей на смену Вакер-процессу (см. ниже), производится более миллиона тонн уксусной кислоты в год.
Кобальт не образует моноядерного карбонила, при взаимодействии порошка металла с монооксидом углерода при повышенном давлении образуются оранжево-красные кристаллы кластерного дикарбонила Co2(CO)8, содержащие две мостиковые СО-группы:
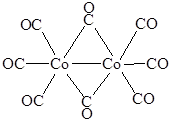
При слабом нагревании вещество превращается в черный тетраядерный кластер Co4(CO)12.
Гидридный комплекс кобальта HCo(CO)4, получаемый при взаимодействии кобальта или карбонила Co2(CO)8 с водородом при высоком давлении СО и повышенной температуре, служит эффективным катализатором гидроформилирования
HCo(CO)4
R2C=CH2 + CO + H2 ¾¾¾¾¾® R2CH–CH2–CHO.
t°, p
Восстанавливая образующиеся альдегиды, получают высшие спирты, используемые во многих типах современных моющих средств.
Родий и иридий также образуют разнообразные кластерные карбонилы, часто содержащие несколько десятков атомов металла, например. [Rh14(CO)25]4–.
ДОПОЛНЕНИЕ. Катализатор Уилкинсона
При кипячении трихлорида родия в этаноле с избытком трифенилфосфина образуются фиолетово-красные кристаллы Rh(PPh3)3Cl. Впервые эта реакция была проведена в 1965 г. английским химиком Дж. Уилкинсоном (в соавторстве с Коттоном он написал несколько учебников по неорганической химии, переведенных на русский язык и широко известных студентам-химикам). Полученное Уилкинсоном вещество устойчиво на воздухе, склонно к реакциям окислительного присоединения, обладает каталитической активностью, например, поджигает водород. Его молекулы имеют плоско-квадратную геометрию (Рис.6.32. Строение катализатора Уилкинсона), в бензольном растворе частично диссоциируют, отщепляя молекулу трифенилфосфина:
Rh(PPh3)3Cl  Rh(PPh3)3Cl + PPh3 , K = 1,4×10–2 (25°C, бензол)
Rh(PPh3)3Cl + PPh3 , K = 1,4×10–2 (25°C, бензол)
При пропускании в этот раствор СО образуется карбонильный комплекс, замещение одной из молекул фосфина происходит также при действии этилена:
В органическом синтезе открытое Уилкинсоном вещество часто используют в качестве активного катализатора гидрирования. Во многих случаях реакции в его присутствии протекают уже при комнатной температуре. Некоторые промышленные установки по гидрированию олефинов также используют катализатор Уилкинсона. Каталитический цикл включает окислительное присоединение водорода, реакции замещения одного из фосфинов алкеном, миграцию координированного алкена и его внедрение по связи Rh-H, восстановительное элиминирование (Рис.6.33. Схема каталитического цикла гидрирования олефинов с помощью катализатора Уилкинсона).
КОНЕЦ ДОПОЛНЕНИЯ
Соединения элементов в степени окисления +2.
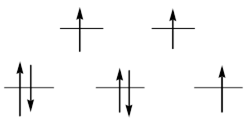
Степень окисления +2 наиболее устойчивая для кобальта. Электронная конфигурация d7, которую он приобретает в этом состоянии, и сравнительно низкая энергия расщепления обеспечивают существование парамагнитных октаэдрических комплексов с частично заполненными как t2g, так и eg-уровнями:
Низкая энергия стабилизации октаэдрическим окружением приводит к энергетической близости между октаэдрическими и тетраэдрическими комплексами, объясняет легкость взаимных переходов между ними. Важную роль в формировании геометрии образующейся комплексной частицы играет стерический фактор: объемные отрицательно заряженные лиганды, такие как Cl–, Br–, I–, SO32–, CO32–, C2O42– способствуют образованию тетраэдрических комплексов, а компактные молекулы H2O, NH3 – октаэрических. В случае некоторых отрицательно заряженных лигандов (OH–, NCS–, F–) в зависимости от условий синтеза могут быть получены комплексы различной геометрии. Часто об изменении координационного числа металла легко судить по переходу одной окраски в другую.
Катион гексааквакобальта(II) [Co(H2O)6]2+ присутствует во многих кристаллических солях, являющихся гексагидратами, например, в сульфате, перхлорате, нитрате, хлориде и бромиде, а также в их разбавленных растворах, которым он придает розовую окраску (Сноска: В водных растворах присутствует также незначительное количество тетраэдрических ионов [Co(H2O)4]2+, находящихся в равновесии с гексааква-формой).
Бледно-розовый осадок гидроксида Co(OH)2 получают приливанием разбавленного раствора нитрата к раствору щелочи, взятой в небольшом избытке. Он изоструктурен гидроксиду железа(II) и состоит из слоев, образованных октаэдрами CoO6, соединенными ребрами (рис.6.34 а). Рис.6.34. Строение гидроксида кобальта Co(OH)2 (а) и основного нитрата Co2(NO3)(OH)3 (б). Изображены слои октаэдров Co(OH)6, атомы кобальта, расположенные в их центре, и атомы водорода не показаны)
Действие щелочи на растворы солей кобальта(II) приводит к образованию синего осадка основных солей, например, Co4(OH)6(SO4) или Co2(OH)3NO3. Они имеют гексагональную слоистую структуру (P. Rabu, S. Angelov, P. Legoll et al, Inorg. Chem., 1993, 32, 2463), отличающуюся от гидроксида тем, что часть гидроксильных групп замещена на анионы, располагающиеся в пространстве между слоями (рис.6.34 б). При добавлении к основным солям избытка щелочи эти анионы вытесняются ионами ОН–, и образуется гидроксид кобальта. Если осаждение гидроксида проводят при 0 °C, приливая раствор соли к раствору щелочи, выделяется синий осадок гидрата 3Co(OH)2×2H2O, в структуре которого молекулы воды занимают пространство между слоями октаэдров. Гидрат легко обезвоживается, превращаясь в розовый осадок Co(OH)2.
Гидроксид кобальта является амфотерным основанием с преобладанием основных свойств (Kb = 4×10–5). С кислотами он дает соли кобальта(II), а растворяясь в концентрированных щелочах при рН > 14, образует темно-синие растворы гидроксокобальтатов [Co(OH)4]2–.
До настоящего времени не описана ни одна кристаллическая структура гидроксокобальтатов(II). В ранней литературе сообщается о синтезе как тетрагидроксо- (Na2[Co(OH)4]), так и гексагидроксо- (Ba2[Co(OH)6]) комплексов. Cиняя окраска ратсворов доказывает преобладание тетраэдрических ионов. При сплавлении оксида или нитрата кобальта(II) с оксидами, гидроксидами и карбонатами металлов в инертной атмосфере получены красные кобальтаты(II), например, Na4CoO3 и Na10Co4O9, в которых содержатся изолированные треугольные ионы CoO34– или объединенные общими вершинами в цепи (Moeller A., Chem. Mater. 1998, 10, 3196; R. Hoppe et al, Z. Anorg. Allg. Chem., 1993, 619, 1807). В соли K2Na2[Co2O5] анион представляет собой два тетраэдра, соединенные общей гранью.
На воздухе розовый осадок гидроксида кобальта(II) постепенно становится коричневым вследствие окисления:
4Co(OH)2 + O2 = 4CoOOH + 2H2O; E°(Co(OH)3/Co(OH)2) = 0.17 B.
Потенциал окисления кобальта(II) в кобальт(III) сильно зависит от кислотности среды, что уже обсуждалось на примере солей железа. Однако, по сравнению с железом(II), соли кобальта(II) устойчивы к окислению кислородом в нейтральной и кислой средах (E°(Co3+/Co2+) = 1.95 B).
Ионы гексааквакобальта(II) в водных растворах частично гидролизованы, однако существующие при этом равновесия практически не исследованы. Отметим образование циклического додекамера [Co12(OH)6(SeO3)8]2+, содержащего мостиковые гидроксогруппы и селенит-ионы (Amoros P., Marcos M.D., Roca M., et al, J. Solid State Chem., 1996, 126, 169) (Рис.6.35. Строение иона [Co12(OH)6(SeO3)8]2+).
Сульфат кобальта(II) («кобальтовый купорос») при комнатной температуре кристаллизуется из водных растворов в форме розово-красного гептагидрата CoSO4×7H2O, изоморфного железному купоросу. При кипячении с концентрированной серной кислотой или при внесении в расплав сульфата аммония он может быть полностью обезвожен. Безводная соль малорастворима в холодной воде, но переводится в раствор кипячением. Она выдерживает нагревание до 800 °C без разложения. При гидролизе могут быть получены основные соли Co4(OH)6SO4, Co5(OH)6(SO4)2×5H2O и Co3(OH)2(SO4)2×2H2O (Dubler E.,Oswald H., Helv. Chim. Acta, 1971, 54, 1621) синего или фиолетового цвета. Известны двойные сульфаты, например (NH4)2Co(SO4)2(H2O)6 - аналог соли Мора.
Сульфит кобальта CoSO3×5H2O получают пропусканием сернистого газа через взвесь гидроксида кобальта. Осаждение сульфитом натрия приводит к основной соли NaCo2(OH)(SO3)2(H2O), а при избытке сульфит-ионов образуются комплексы, например, Na2[Co(SO3)2].
Нитрат кобальта(II) Co(NO3)2 кристаллизуется в виде красных призматических кристаллов гексагидрата из растворов, полученных обработкой карбоната или гидроксида азотной кислотой. При 55 °C соль плавится, при 60 °C теряет половину кристаллизационной воды, а затем разлагается с образованием Со3О4. Безводный нитрат получают взаимодействием порошка кобальта с N2O4 или обезвоживанием азотнокислого раствора нитрата в эксикаторе над фосфорным ангидридом (G.A. Tikhomirov, K.O. Znamenkov, I.V. Morozov, et al, Z. Anorg. Allg. Chem., 2002, 628, 269).
Средний ортофосфат кобальта Co3(PO4)2×8H2O получают с использованием гидрофосфата натрия:
3CoSO4+ 4Na2HPO4 = Co3(PO4)2¯ + 2NaH2PO4 + 3Na2SO4.
При 900 °C вещество теряет воду, превращаясь в фиолетовый порошок, используемый в качестве пигмента («темно-фиолетовый кобальт»). Если осаждение проводить с помощью (NH4)2HPO4, образуется осадок двойной соли CoNH4PO4×H2O (пигмент «светло-фиолетовый кобальт»). Средний ортофосфат осаждает из растворов солей кобальта темно-фиолетовые основные соли Co5(PO4)2(OH)4 и Co2OHPO4. Известен кислый фосфат CoHPO4×H2O, Осадки фосфатов кобальта растворимы в растворах аммиака, а также в кислотах – даже таких слабых, как уксусная. Под действием фосфорной кислоты они переходят в Co(H2PO4)2. Получена кислая соль Co3(HPO4)2(OH)2 содержащая ОН-группы (Pizarro J.L., Villeneuve G., Hagenmuller P., Le Bail A., J. Solid State Chem., 1991, 92, 273). Известны фосфит CoHPO3(H2O) и гипофосфит Co(H2PO2)2(H2O)6 кобальта.
Средний карбонат кобальта(II) нельзя осадить из раствора карбонатом натрия вследствие образования синей основной соли. Его получают действием на водный раствор нитрата гидрокарбонатом натрия, насыщенным углекислым газом. На холоду образуется гексагидрат розово-фиолетового цвета, при нагревании в запаянной трубке до 140 °C он полностью обезвоживается, при 400 °C разлагается
При действии на основную или среднюю соль избытком раствора карбоната щелочного металла или аммония образуется ярко-красный комплексный карбонат M2[Co(CO3)2], малорастворимый в воде.
Розово-красный оксалат кобальта CoC2O4×2H2O получают по реакции обмена. Он нерастворим в воде, при нагревании в инертной атмосфере разлагается на пирофорный кобальт и углекислый газ. В присутствии оксалат-ионов дает комплексы.
Ацетат кобальта кристаллизуется из водных растворов в виде тетрагидрата, который при 140 °C теряет кристаллизационную воду, а при 270 °C разлагается на оксосоль Co3O(CH3COO)4, углекислый газ и ацетон.
При помещении раствора нитрата кобальта в трифтоуксусной кислоте в эксикатор с фосфорным ангидридом получены кристаллы (NO)[Co3F(CF3COO)6(CF3COOH)3], содержащие трехъядерный анионный комплекс, строение которого напоминает широко известные оксоацетаты металлов(III). В центре комплекса расположен ион F–, координирующий сразу три атома кобальта (Рис.6.36 Строение аниона [М3F(CF3COO)6(CF3COOH)3]–, M = Co, Ni. Атомы фтора трифторметильных групп не показаны). Шесть трифторацетатных групп связывают атомы металла в цикл, а три молекулы трифторуксусной кислоты выполняют функцию монодентатных лигандов. Аналогичное соединение получено и для никеля (Д.С. Терещенко, И.В. Морозов, А.И. Болталин, Е. Кемниц, С.И. Троянов, Журн. неорган. хим., 2004, 49, 836).
Комплексные соединения кобальта(II) лабильны и характеризуются сравнительно низкими значениями констант устойчивости, и поэтому легко вступают в реакции замещения. Одновременное присутствие в растворе двух лигандов, один из которых имеет тенденцию к образованию тетраэдрических комплексов, а другой – октаэдрических, приводит к равновесию, которым легко управлять, изменяя концентрацию реагентов или температуру. Так, при добавлении соляной кислоты к розовому раствору хлорида кобальта окраска становится синей из-за образования хлоридного комплекса:
[Co(H2O)6]2+ + 4Cl–  [CoCl4]2– + 6H2O.
[CoCl4]2– + 6H2O.
розовый синий
При разбавлении водой раствор вновь приобретает розовый цвет, а последующее нагревание, а также добавление спирта или ацетона опять приводит к появлению синей окраски (Рис.6.37. Электронный спектр поглощения растворов, содержащих 0,1М [Co(H2O)6]2+ и 0,001М [CoCl4]2–). Таким образом, образованию хлоридного комплекса способствует избыток хлорид-ионов, повышение температуры, замещение воды на органический растворитель либо ее удаление из сферы реакции. Поэтому слово, написанное водным растором хлорида кобальта и практически не заметное на бумаге, выступает синими буквами при нагревании. Папиросная бумага, пропитанная таким раствором, в сухую погоду имеет розовый цвет, а в мокрую – синий. Гораздо более интенсивный цвет тетраэдрического комплекса объясняется отсутствием центра симметрии в тетраэдре, что увеличивает вероятность d-d переходов, а разница в окраске обусловлена различным характером расщепления и величинами ЭСКП.
Такое же обратимое изменение окраски происходит при добавлении к растворам солей кобальта ледяной уксусной кислоты, роданида аммония. Все эти превращения свидетельствуют о лабильности комплексов кобальта(II).
Бромидные и иодидные комплексы по строению и свойствам напоминают хлоридные. Со фторид-ионами, помимо комплексов с координационным числом кобальта четыре (K[CoF3],K2[CoF4], K3[Co2F7]), известен также гексафторид (NH4)4[CoF6], осаждаемый фторидом аммония из спиртового раствора хлорида кобальта(II) в отсутствие воды. В водном растворе комплекс неустойчив. Из-за больших ионных радиусов хлорид-, бромид- и иодид-ионов, они образуют с кобальтом лишь тетраэдрические комплексы. Даже соль сосава (NH4)3CoCl5 состоит из ионов аммония, тетраэдров [CoCl4]2– и хлорид-ионов Cl– (Рис. 6.38. Строение (NH4)3[CoCl4]Cl. Атомы водорода ионов аммония не показаны).
Многие комплексные соединения кобальта(II) легко могут быть окислены до соединений кобальта(III). Это сопровождается удалением электрона с разрыхляющей eg-орбитали и увеличением ЭСКП. Таким образом, процесс является термодинамически благоприятным. Чем сильнее кристаллическое поле лиганда, тем выше восстановительная способность комплекса кобальта(II) (Рис.6.39. Зависимость величины стандартного элеткродного потенциала Co(III)/Co(II) от природы лиганда)
Амминокомплексы образуются при добавлении избытка аммиака к раствору соли кобальта(II). Выпадающий сначала синий осадок основного хлорида при дальнейшем прибавлении аммиака растворяется, образуя грязно-желтый раствор. Для получения кристаллической соли гексаамминкобальта проводят взаимодействие безводного галогенида с газообразным аммиаком:
CoCl2 + 6NH3 = [Co(NH3)6]Cl2
Аммиакат обладает сравнительно низкой устойчивостью (Kуст = 2,4×104), и при растворении в воде часть молекул аммиака замещается на воду. Подобно большинству других комплексов кобальта(II) он неустойчив к окислению, на воздухе превращаясь в аммиачные комплексы кобальта(III).
Ацетилацетонат кобальта выпадает в виде розово-красного дигидрата при действии ацетилацетона и основания на растворы солей кобальта(II). При нагревании он теряет воду и сублимируется, преврашаясь в тетрамерные молекулы Co4(acac)8, устойчивые не только в кристаллах, но и в паре. Сходными свойствами обладает и ацетилацетонат никеля, который, однако, представляет собой тример Ni3(acac)6 (Рис.6.40 Строение ацетилацетонатов кобальта и никеля: (a) M(acac)2(H2O)2, M = Co, Ni, (б) Co(acac)3, (в) Co4(acac)8, (г) Ni3(acac)6). Летучесть дикетонатных комплексов металлов используется не только для их очистки сублимацией, но и для нанесения оксидных и металлических покрытий методом осаждения из газовой фазы.
При действии цианидом калия на растворы солей кобальта(II) образуются зеленые растворы, из которых кристаллизуются пурпурные соли, содержащие диамагнитный ион [Co2(CN)10]6– с терминальными цианидными групами и связью металл-металл. В растворе он расщепляется на два пентацианата, а шестое место в координационной сфере занимает молекула воды. Пентацианоаквакобальтат(II) [Co(CN)5(H2O)]3– принадлежит к числу немногочисленных низкоспиновых октаэдрических комплексов кобальта(II) с конфигурацией t2g6eg1. Потеря единственного электрона, расположенного на разрыхляющей eg-орбитали, настолько благоприятна энергетически, что комплекс является очень сильным восстановителем – он медленно разлагает воду с выделением водорода:
2[Co(CN)5(H2O)]3– + 2CN– = 2[Co(CN)6]4– + 2OH– + H2.
В ацетонитрильном растворе пентацианокобальтат способен поглощать водород, восстанавливая его до гидридного комплекса кобальта(III):
2[Co(CN)5]3– + H2 = 2[Co(CN)5Н]3–
и кислород, превращаясь в супероксокомплекс:
2[Co(CN)5]3– + О2 = 2[Co(CN)5О2]3–
Соединения в степени окисления +2, наиболее устойчивой у кобальта, для родия и особенно иридия, напротив, не характерны. Исходя из электронной конфигурации d7, следовало бы ожидать, что они будут парамагнетиками, по аналогии с соединениями кобальта. Примеров таких комплексов крайне мало, например, неустойчивый циклопентадиенильный комплекс – родоцен Rh(C5H5)2. Для родия(II) наиболее изучены диядерные карбоксилаты, построенные аналогично ацетатам хрома(II) и молибдена(II).
При кипячении оксида родия(III) или трихлорида с уксусной кислотой в этаноле раствор приобретает зеленый цвет, а при охлаждении из него кристаллизуется сине-зеленые кристаллы ацетата Rh2(CH3COO)4. Диамагнетизм комплекса доказывает наличие сильного взаимодействия между атомами родия. Вещество легко присоединяет нейтральные лиганды L, которые занимают вакантные позиции у атомов металла: Rh2(CH3COO)4L2. Таким путем могут быть получены гидрат, комплексы со спиртами и фосфинами. Соединения с кислород-донорными лигандами имеют сине-зеленую окраску, а с фосфинами – оранжево-красную, что свидетельствует о π-связывании. При обработке ацетата тетрафтороборной кислотой удается вытеснить ацетатные группы из координационной сферы родия. Образующиеся зеленые растворы содержат сольватированные катионы Rh24+, однако, в твердом виде такие соли выделить пока не удалось. Мостиковую функцию, аналогичную ацетатным группам, способны выполнять и сульфат и карбонат-ионы: известны комплексы Na4[Rh2(SO4)4(H2O)2] (Дикарева Л.М., Зефиров А.Н. Жилаев А.Н., Барановский И. Б., Порай-Кошиц М.А., Журн. неорг. хим., 1987, 32, 64 АНГЛ) и Na4[Rh2(СO3)4] (Wilson C.R., Taube H., Inorg. Chem., 1974, 14, 405), образующиеся из ацетата по обменным реакциям. Интересно, что ближайший аналог родия иридий(II) не образует диядерных карбоксилатов.
ДОПОЛНЕНИЕ. Кобаламины
Кобаламинами называют комплексы кобальта с корриноидами – природными порфиринами, содержащими цикл коррина. Наиболее интересны биологически активные кобаламины, такие как метилкобаламин, цианокобаламин (витамин В12) и 5’-дезоксиаденозилкобаламин (кофермент В12). Они представляют собой низкоспиновые октаэдрические комплексы кобальта(III), в которых в качестве лиганда выступает корриноид, содержащий сложный органический заместитель, завершающийся бензимидазольной группой (Рис. (а) Цикл коррина, (б) Структура витамина В12). За определение кристаллической структуры витамина В12 английский химик Д. Кроуфут-Ходжкин был удостоен в 1964 г. Нобелевской премии. Он доказал, что атом кобальта в комплексе координирован четырьмя атомами азота порфиринового цикла, атомом азота бензимидазола и органическим заместителем R, различным для разных кобаламинов. Благодаря легкости разрыва связи Со-R по радикальному механизму, кобаламины обладают способностью переноса метильных групп от одной органической молекулы к другой. Например, в организме они катализизируют изомеризацию метилмалоновой кислоты в янтарную, синтез метионина из гомоцистеина путем переноса на него метильной группы:

Кофермент В12 участвует в переносе ионов водорода, при этом последовательно восстанавливаясь:
В организме человека содержится около пяти миллиграммов кобаламинов, поступающих с животной пищей или вырабатываемых микрофлорой кишечника. В качестве медицинского препарата используют цианокобаламин – рубиново-красные кристаллы, растворимые в воде и спирте, неустойчивые к действию света. В организме он легко превращается в другие биологически активные формы.
КОНЕЦ ДОПОЛНЕНИЯ
Соединения в степени окисления +3.
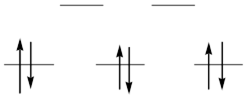
Степень окисления +3 соответствует электронной конфигурации d6, которая оказывается наиболее устойчивой как термодинамически, так и кинетически в низкоспиновых октаэдрических комплексах (t2g6):
Именно поэтому для родия, у которого в силу высокой ЭСКП все комплексы низкоспиновые, данная степень окисления оказывается наиболее устойчивой, в то время как для кобальта ее стабилизируют лишь лиганды сильного и среднего поля. В то же время, в случае лигандов слабого поля, с которыми кобальт(III) вынужден образовывать высокоспиновые комплексы, данная степень окисления оказывается нестабильной. Именно поэтому простые соли кобальта(III) неустойчивы и являются сильными окислителями.
Недавние исследования доказали, что ион [Co(H2O)6]3+ является диамагнитным, то есть низкоспиновым. Это единственный пример низкоспинового аквакомплекса среди 3d-металлов. Его образование объясняется энергетическим выигрышем при заполнении электронами t2g-орбиталей и высоким значением параметра расщепления Δo. Фторидные комплексы кобальта(III) [CoF6]3– и [CoF3(H2O)3] - высокоспиновые. Гексафторокобальтат(III) калия K3CoF6 образуется при фторировании смеси хлоридов кобальта(II) и калия при 400 °C. Они представляют собой светло-желтые порошки, разлагающиеся водой.
Ионы [Co(H2O)6]3+ могут быть получены электрохимическим окислением растворов перхлората кобальта(II) или подкислением зеленых растворов, образующихся при действии на соли кобальта(II) пероксида водорода в насыщенном растворе гидрокарбоната натрия.
Разработанный недавно (G. Wangila, R.B. Jordan, Inorg. Chim. Acta, 2003, 343, 347) удобный способ приготовления водных растворов, содержащих, заключается в действии 2М хлорной кислоты на зеленую соль [Co(NH3)6][Co(CO3)3]. Ионы водорода разлагают карбонатный комплекс, а аммиакат выделяется из сферы реакции в форме желтого осадка перхлората:
[Co(NH3)6][Co(CO3)3] + 6H+ + 6ClO4– = [Co(H2O)6]3+ + [Co(NH3)6](ClO4)3¯ + 3CO2
зеленый синий желтый
Исходный комлпекс легко приготовить из [Co(NH3)6]Cl3 и зеленых растворов, полученных окислением нирата кобальта(II) пероксидом водорода в насыщенном растворе NaHCO3.
Растворы, содержащие ионы гексааквакобальта(III), имеют синюю окраску. Они очень
неустойчивы и разлагаются с выделением кислорода:
4[Co(H2O)6]3+ + 2H2O = 4[Co(H2O)6]2+ + 4H+ + O2
Период полураспада составляет около месяца.
Из солей кобальта(III) в кристаллическом виде получены сульфат и фторид. Синие пластинчатые кристаллы Co2(SO4)3×18H2O образуются при анодном окислении сернокислого раствора сульфата кобальта(II) и последующем охлаждении до 0 °C. В качестве окислителя возможно использование фтора. При добавлении сульфата щелочного металла кристаллизуются темно-синие квасцы CsCo(SO4)2×12H2O.
При электролизе раствора соли кобальта в 40%-ной плавиковой кислоте может быть получен зеленый осадок фторида CoF3×4.5H2O. Взаимодействием безводного фторида с азотным ангидридом получают нитрат Co(NO3)3. Все эти соли разлагаются водой и являются сильными окислителями.
При окислении ацетата кобальта(II) образуется смесь различных оксо- и гидроксоацетатов кобальта(III), содержащая димер Co2(OH)2(CH3COO)4 с мостиковыми ОН-группами и трехъядерный оксоацетат [Co3O(CH3COO)6(H2O)3]CH3COO, построенный аналогично оксоацетатам других трехзарядных катионов. Средний фосфат кобальта(III) неизвестен, но описаны аммиачные комплексы CoPO4(NH3)5(H2O)2 и CoPO4(NH3)4(H2O)3 (Solans X., Rius J., Miravitlles C., Z. Kristallogr., 1981, 157, 207).
При действии щелочи на растворы, содержащие ионы гексааквакобальта(III), выпадает коричневый осадок гидроксида переменного состава, который при хранении или небольшом нагревании превращается в устойчивую оксогидроксоформу СоООН, соответствующую природному минералу гетерогениту. В его структуре октаэры CoO6 сочленены общими ребрами в слои, объединенные друг с другом водородными связями (Рис.6.42. Строение СоООН). Осадок оксогидроксида не растворим в серной кислоте, но в смеси ее с пероксидом водорода
или нитритом натрия, восстанавливающими кобальт(III) до соли кобальта(II), переходит в раствор:
2CoOOH + 2H2SO4 + H2O2 = 2CoSO4 + O2 + 4H2O (Сноска: в щелочной среде пероксид водорода окисляет кобальт(II) до кобальта(III))
При сплавлении с щелочами он дает кобальтаты(III), например, NaCoO2, Na5CoO4 (Сноска: в структуре Na5CoO4 присутствуют изолированные тетраэдры СоО4, что не свойственно кобальту(III)), а при нагревании превращается в шпинель Со3О4.
Для кобальта(III) характерны октаэдрические низкоспиновые комплексы. Наличие полностью заселенных t2g-орбиталей делает их химически инертными и усложняет синтез по реакциям обмена. Общий метод получения комплексных соединений кобальта(III) заключается в окислении солей кобальта(II) в присутствии лиганда. Так, при пропускании воздуха через аммиачный раствор хлорида кобальта(II) появляется красное окрашивание, вызванное образованием аммиаката кобальта(III):
4CoCl2 + 16NH3 + 4NH4Cl + O2 = 4[Co(NH3)5Cl]Cl2 + 2H2O.
Окисление протекает в несколько стадий через образование коричневого интермедиата с мостиковой пероксо-группой, который может быть выделен в свободном виде.
Это вещество – наиболее известный диядерный пероксидный комплекс кобальта. В кислой среде он восстанавливается до соли кобальта(II), выделяя кислород, а под действием сильных окислителей приобретает один электрон и превращается в зеленый супероксокомплекс кобальта(III), в котором в качестве лиганда выступает супероксогруппа О2– (Рис.6.43. Схема синтеза некоторых аммиакатов кобальта). Получены также диядерные аммиакаты, содержащие две и даже три мостиковые гидроксо-группы Thewalt U., Z. Anorg. Allg. Chem., 1975, 412, 29; W. Frank, Hoffman K., Heck L., Angew. Chem., Int. Ed. Engl., 1990, 29, 1158):

В присутствии активированного угля при окислении аммиачного комплекса кобальта(II) образуется гексаммин, выпадающий в виде винно-красных кристаллов:
уголь
4CoCl2 + 20NH3 + 4NH4Cl + O2 = 4[Co(NH3)6]Cl3 + 2H2O.
По сравнению с аналогичным комплексом кобальта(II), ион [Co(NH3)6]3+ обладает высокой термодинамической (Куст = 1.62×1035) и кинетической устойчивостью. Его часто используют для кристаллизации трехзарядных анионов больших размеров. Так, в виде солей с катионом [Co(NH3)6]3+ получены твердые хлоридные комплексы многих металлов: [MCl6]3– (M(III) = Fe, Sb, Bi), [MCl5]2– (M(II) = Hg, Cd, Cu), [Sb2F9]3–, [Pb4Cl11]3– , а также цианидные, карбонатные и многие другие комплексные соли. Чем больше размер аниона, тем менее растворим комплекс. На этом, например, основан метод получения оксалата или иодида:
[Co(NH3)6]Cl3 + 3KI = [Co(NH3)6]I3¯ + 3KCl.
Разложение комплексов кислотами предполагает первоначальное протонирование уходящей группы. В случае катиона [Co(NH3)6]3+ неподеленные пары молекул аммиака участвуют в образовании связей с атомом кобальта, поэтому комплекс оказывается очень устойчивым к действию даже концентрированных кислот – серной и азотной! Он может быть разрушен лишь горячими концентрированными растворами щелочей, которые вызывают депротонирование одной из молекул аммиака, приводящее к образованию интермедиата с координационным числом пять, стабилизированного π-связыванием.
Тетраммины кобальта(III) получают, вводя в синтез наряду с аммиаком карбонат-ионы –бидентатные лиганды, которые легко удаляются протонированием:
4CoSO4 + 4(NH4)2CO3 + 8NH3 + O2 = 4[Co(NH3)4CO3]SO4 + 2H2O.
При охлаждении из раствора выделяются гранатово-красные призматические кристаллы. Обработка их раствором серной кислоты приводит к цис-диакватетраммину [Co(NH3)4(Н2О)2]SO4, из которого последовательной обработкой концентрированной серной и соляной кислотами получают зеленые кристаллы транс-[Co(NH3)4Cl2]Cl.
Оранжево-желтые игольчатые кристаллы хлорида трис(этилендиамин)кобальта(III) образуются при окислении хлорида кобальта кислородом воздуха или пероксидом водорода при рН, близком к нейтральному:
4CoCl2 + 8en + 4en×HCl + O2 = 4[Co(en)3]Cl3 + 2H2O.
Соль представляет собой смесь двух оптических изомеров

которые могут быть разделены кристаллизацией из растворов, содержащих оптически активную соль винной кислоты H2tart(+). Правовращающий изомер [(+)Co(en)3][(+)tart]Cl×5H2O кристаллизуется из раствора в форме темно-оранжевых кристаллов, а левовращающий остается в растворе. При кипячении раствора происходит рацемизация. Метод разделения оптически активных изомеров был впервые разработан Альфредом Вернером в 1911 г. как раз на примере комплексов кобальта с этилендиамином. Современные исследования вернеровских комплексов опубликованы в: W.G. Jackson, J.A: McKeon, S. Cortez, Inorg. Chem., 2004, 43, 6249.
Если окисление хлорида кобальта в водном растворе проводить в солянокислой среде, то образуется зеленый транс-изомер бис-этилендиаминового комплекса [H5O2][Co(en)2Cl2]Cl2. При слабом нагревании в вакууме он отщепляет воду и хлороводород без изменений в координационной сфере кобальта:
60 °С, вакуум
[H5O2][Co(en)2Cl2]Cl2 ¾¾¾¾¾¾® транс-[Co(en)2Cl2]Cl + HCl + 2H2O.
Кипячение водного раствора сопровождается изменением окраски из зеленой в красно-фиолетовую, что объясняется изомеризацией:
100 °C
транс-[Co(en)2Cl2]+ ¾® цис-[Co(en)2Cl2]+
зеленый красно-фиолетовый
а также замещением одного из атомов хлора на молекулу воды
транс-[Co(en)2Cl2]+ + H2O ¾® [Co(en)2Cl(H2O)]2+ + Cl– (28 % цис и 72 % транс).
Атомы хлора в бис-этилендиаминовых комплексах могут быть легко замещены на другие галогены и псевдогалогены, например, роданид, азид.
ДОПОЛНЕНИЕ. Нитро- и нитрито комплексы – пример изомерии лиганда.
Нитрит-ион имеет два типа донорных атомов – азот и кислород, поэтому может координироваться через любой из них в зависимости от природы комплексообразователя. С мягкими кислотами Пирсона образуются нитро-комплексы, содержащие связь металл-азот, а с жесткими – нитрито-комплексы, в которых металл связан с атомом кислорода. Для кобальта(III) известны оба типа координации, хотя более устойчивы нитро-комплексы.
При кипячении раствора соли кобальта, подкисленного уксусной кислотой, с избытком нитрита калия выпадает желтый осадок гексанитрокобальтата калия:
Co(NO3)2 + 7KNO2 + 2CH3COOH = K3[Co(NO2)6]¯ + NO + 2CH3COOK + 2KNO3 + H2O.
В некоторых учебных пособиях это вещество ошибочно называют нитрито-комплексом. В его структуре присутствуют ионы с геометрией правильного октаэдра (расстояния Со-N 0,198 нм (Рис.6.44. Строение [Co(NO2)6]3–) (Громилов С.А., Алексеев В.И. и др., Журн. неорган. хим., 1992, 37, 615). Изомерный ему гексанитритокомплекс не получен.
Натриевую соль получают по аналогичной реакции, но выделяют из раствора
высаливанием спиртом, так как она хорошо растворима в воде. Растворимость гексанитритокобальтатов(III) щелочных металлов, таким образом, понижается с ростом радиуса катиона. Натриевую соль используют в качестве реагента на ионы калия.
Взаимные переходы между нитрито и нитро-измерами удобно изучать на пентамминах.
Розово-красный осадок нитритного комплекса образуется при замещении атома хлора в ионе пентамминкобальта(III) на нитритную группу:
[Co(NH3)5Cl]Cl2 + NaNO2 = [Co(NH3)5(ONO)]Cl2 + NaCl.
В слабокислой среде он превращается в желтый нитро-комплекс:
H+
[Co(NH3)5(ONO)]Cl2 ¾¾¾® [Co(NH3)5(NO2)]Cl2
розово-красный желтый
нитрито нитро
В твердом соединении изомеризация происходит спонтанно, но за более длительное
время – в течение нескольких недель (константа скорости 10–4 с–1 при 58 °C). Обратный переход возможен лишь при фотохимической активации.
Под действием кислот нитритный комплекс разрушается.
КОНЕЦ ДОПОЛНЕНИЯ.
Среди комплексов с хелатирующими кислород-донорными лигандами наиболее
известны зеленый ацетилацетонат Co(acac)3 (рис.6.40 б) и трисоксалатокобальтат(III) K3[Co(C2O4)3], образующийся при окислении трис-оксалатного комплекса кобальта(II).
Степень окисления +3 является наиболее устойчивой для родия. При взаимодействии с
кислотами оксида родия(III) образуются желтые растворы, содержащие диамагнитные (t2g6) ионы [Rh(H2O)6]3+. Из них кристаллизуются соли, например, перхлорат Rh(ClO4)3×6H2O, сульфат Rh2(SO4)3×18H2O или квасцы CsRh(SO4)2×12H2O. При выпаривании раствора сульфата, подкисленного серной кислотой, выделяются красные кристаллы состава, по-видимому, представляющие собой комплекснyю кислоту, как и нитрат Rh(NO3)3×2H2O, также имеющий красную окраску. Строение этих веществ неизвестно.
Соли родия(III) в водных растворах сильно гидролизованы (I. Banyai, et al, Inorg. Chem.,
1995, 34, 2423):
[Rh(H2O)6]3+  [Rh(H2O)5(OH)]2+ + H+ , K = 2.5×10–4
[Rh(H2O)5(OH)]2+ + H+ , K = 2.5×10–4
В продуктах их гидролиза присутствуют диядерные гидроксокомплексы
[(H2O)4Rh(μ-OH)2Rh(H2O)4]4+.
Комплексы родия(III) во многом напоминают комплексы кобальта(III): они
преимущественно октаэдрические и низкоспиновые, даже с лигандами слабого поля, например, водой и фторидом (с этими лигандами кобальт(III) дает высокоспиновые комплексы). Все они диамагнитны и практически не проявляют окислительных свойств. Сходство с кобальтом распространяется и на важнейшие типы комплексных соединений, например, пента- и гексаммины, цианидные и оксалатные комплексы. Известны также и мостиковые супероксо-димеры. Катионные и нейтральные комплексы родия(III), подобно комплексам кобальта(III), химически инертны, а анионные комплексы родия, в отличие от них, лабильны.
В химии родия(III) важную роль играют комплексные хлориды. Гексахлорородат(III)
натрия Na3RhCl6 получают нагреванием смеси безводного трихлорида родия и хлорида натрия в токе хлора при 300 °C. При растворении в воде и упаривании раствора вещество выделяется в виде гидрата интенсивно красного цвета.
При действии щелочи на водные растворы гексахлорородатов или солей родия(III)
выделяется желтый осадок гидратированного оксида примерного состава Rh2O3×5H2O. Он растворяется в кислотах, а с щелочами реагирует лишь при сплавлении, образуя родиты, например, NaRhO2 (Hobbie K., Hoppe R., Z. Anorg. Allg. Chem., 1988, 565, 106) или MgRh2O4 со структурой шпинели.
Удобным исходным соединением для синтеза соединений родия является
трихлорид, кристаллизующийся из растворов, полученных растворением Rh2O3×5H2O в минимальном количестве концентрированной соляной кислоты. Состав выделяющихся темно-красных кристаллов примерно соответствует тригидрату RhCl3×3H2O. В воде он образует розовые растворы, что объясняет название, данное элементу, которое происходит от греческого ροδον – роза. Их окраска обусловлена образованием хлоридных комплексов [RhCln(H2O)6–n](3–n)+.
При взаимодействии трихлорида родия с раствором оксалата образуются оксалатные комплексы,
например, K3[Rh(C2O4)3], существующий в форме двух оптических изомеров, а с раствором нитрита – нитритные, например, Li3[Rh(NO2)6] (Громилов С.А., Байдина И.А. и др., Журн. неорган. хим., 1994, 39, 109). При сплавлении этого соединения с кислым фторидом щелочного металла может быть получен гексафторородат(III):
K3[Rh(NO2)6] + 6KHF2 = K3[RhF6] + 3NO + 3NO2 + 3H2O.
При нагревании раствора трихлорида родия с аммиаком и хлоридом аммония образуется светло-желтый
осадок хлоропентамминродия(III)
RhCl3×3H2O + 5NH3 = [RhCl(NH3)5]Cl2 + 3H2O.
При нагревании его с водным раствором аммиака в запаянной ампуле могут быть получены бесцветные
кристаллы гексаммина [Rh(NH3)6]Cl3. Среди комплексов с хелатирующими лигандами упомянем ацетилацетонат, состоящий из молекул Rh(acac)3.
Угарный газ, а также органические вещества, содержащие карбонильную группу, восстанавливают
трихлорид родия в водном растворе до карбонильных комплексов родия(I). Такая реакция, например, протекает с муравьиной кислотой или диметилформамидом (Сноска: Кипячением K3[IrCl6] с трифенилфосфином в диметилформамиде получают катализатор Васка Ir(CO)(PPh3)2Cl). А при взаимодейсттвии трихлорида родия с цинком в аммиачном растворе и в присутствии сульфат-ионов восстановление родия не происходит, а образуется гидридный комплекс родия(III) [RhH(NH3)5]SO4 (K. Lemma, A. Bakac, Inorg. Chem., 2004, 43, 4505).
Желтый димагнитный гексаакваион иридия(III) может быть получен при
растворении гидратированного оксида в хлорной кислоте, в твердом виде он известен в квасцах CsIr(SO4)2×12H2O (Armstrong R.S., Beattie J.H., Best S.P., Skelton B.W., White A.H., Journ. Chem. Soc., Dalton Trans., 1983, 1973). Ион [Ir(H2O)6]3+ является рекордсменом по инертности – обмен координированной воды происходит за сотни лет! При действии щелочей на растворы солей иридия(III) образуется зеленый осадок гидратированного оксида Ir2O3×xH2O, растворимый не только в кислотах, но и в щелочах с образованием гексагидроксоиридатов(III) [Ir(OH)6]3–. Соли иридия(III), в отличие от аналогичных соединений кобальта и родия, на воздухе легко окисляются до иридия(IV). Их получают восстановлением иридия(IV) оксалатом натрия, взятом в стехиометрическом количестве, или сахарозой:
2Na2[IrCl6] + Na2C2O4 = 2Na3[IrCl6] + 2CO2.
При аффинаже под действием царской водки иридий частично переходит в раствор в форме гексахлороиридиевой(IV) кислоты. Для отделения от гексахлороплатината раствор до добавления ионов аммония восстанавливают сахарозой, при этом иридий восстанавливается до H3[IrCl6]. При действии хлорида аммония платина осаждается в форме (NH4)2PtCl6, а иридий остается в растворе. Действием на Na3[IrCl6] оксалатом калия получают K3[Ir(C2O4)3].
Трихлорид иридия образует гидрат переменного состава, представляющий собой
смесь различных хлороаквакомплексов (G. Wilkinson et al, Journ. Chem. Soc., Dalton Trans., 1993, 3219) – именно они придают его водным растворам грязно-зеленый цвет. Синтез и свойства пента- и гексааммиакатов иридия(III) аналогичны описанным выше для родия(III). Оба металла образуют оксиацетаты [M3O(CH3COO)6(H2O)3]CH3COO, построенные идентично аналогичным соединениям алюминия, хрома(III), марганца(III), железа(III) и кобальта(III). При восстановлении гидратированного диоксида хлороводородом получены темно-зеленые гигроскопичные кристаллы гидроксохлорида Ir(OH)Cl2, легко растворимые в воде с образованием темно-зеленых растворов.
Соединения кобальта, родия и иридия в высших степенях окисления
Устойчивость высших степеней окисления возрастает вниз по группе. Если
соединения кобальта(IV) и родия(IV) неустойчивы и проявляют свойства сильных окислителей, то степень окисления +4 для иридия в некоторых случаях оказывается наиболее стабильной.
Черный порошок гидратированного диоксида CoO2×xH2O считают конечным
продуктом окисления солей кобальта в щелочных растворах гипохлоритом или озоном, однако это вещество до сих пор не изучено. Красно-коричневые кобальтаты(IV), полученные окислением надпероксидами или кислородом:
1000 °C
12NaO2 + Co3O4 ¾¾®3Na4CoO4 + 8O2
1050 °C
2Ba(OH)2 + Co(OH)2 + O2 ¾¾®Ba2CoO4 + 3H2O,
состоят из отдельных тетраэдров СоО4 или из октаэдров СоО6, связанных общими ребрами в бесконечные цепи.
Кобальт(IV) может быть стабилизирован во фторидe Cs2CoF6, гетерополисоединении
K6CoMo9O32×6H2O, образующемся при окислении соли кобальта(II) персульфатом в присутствии гептамолибдата, а также в комплексах с дитиокарбаматами и макроциклическими лигандами.
Соединения родия в высших степенях окисления немногочисленны. Многие
соединения, образующиеся при окислении щелочных растворов комплексов родия(III), как доказано в настоящее время, являются комплексами родия(III). Например, при образовании клауссовой сини, осуществляемым пропусканием хлора через щелочной раствор трихлорида родия, имеет место окисление двух гидроксо-групп в O22–, а степень окисления родия остается неизменной:
2RhCl3 + Cl2 + 5NaOH + 3H2O = [Rh2(OH)2(H2O)4(μ-O2)]Cl3 + 5NaCl.
Синь представляет собой, таким образом, супероксокомлпекс родия(III) (I.J. Ellison, ZR.D. Gillard, M. Moszner et al, Journ. Chem. Soc., Dalton Trans., 1994, 2531).
Среди немногих достоверных соединений родия(IV) известны хлоридные комплексы, например, зеленый Cs2[RhCl6], образующийся при окислении гексахлорородата(III) сульфатом церия(IV) и хлором. Водой он разлагается с выделением хлора. При сплавлении порошка родия со щелочами могут быть получены родаты, например, Na2RhO3. Химия родия(V) представлена фторидом RhF5 и фторородатами(V), например, CsRhF6.
Для иридия степень окисления +4 более устойчива, чем у других элементов группы. Окисление гексааква-иона иридия(III) электрохимически или солями церия(IV) приводит к зеленовато-коричневым растворам соединений иридия(V), которые в течение нескольких часов, окисляя воду, становятся сине-фиолетовыми – в них содержатся катионы иридия(IV) [(H2O)4Ir(μ-OH)2Ir(H2O)4]6+ и [(H2O)4Ir–O–Ir(H2O)4]6+, а затем желтыми, в результате превращения в соли иридия(III) (D.T. Richens, Inorg. Chem., 1989, 28, 954). В твердом виде солей иридия(IV) с кислород-содержащими анионами выделить не удалось. Окисление оксиацетата иридия(III) озоном также приводит к синим растворам, в которых часть атомов иридия в триядерных катионах окислена до +4 (A. Bino, Inorg. Chim. Acta, 1993, 213, 99). Комплексные соединения иридия(IV) получены и в твердом виде. Так, окисление оксалатного комплекса иридия(III) хлором приводит к Cs2[IrCl4(C2O4)], а аммиаката – к [Ir(NH3)4Cl2]Cl2.
Наиболее устойчивы галогенидные комплексы иридия(IV), например, гексахлороиридат(IV), образущийся при хлорировании смеси порошка иридия с хлоридом калия:
625 °C
2KCl + Ir + 2Cl2 ¾¾® K2IrCl6
Соль представляет собой темно-красные кристаллы, растворимые в воде. По реакциям обмена может быть получена и иридийхлороводородная кислота Н2IrCl6, кристаллизующаяся из водных или спиртовых растворов в виде черно-красных игольчатых кристаллов.
Действием щелочи на хлороиридаты(IV) получают черный осадок гидратированного оксида IrO2×2H2O, легко растворимый в соляной кислоте. При 350 °C он полностью обезвоживается, превращаясь в оксид IrO2. Сплавлением его со щелочами получены иридаты(IV), например, Na2IrO3, CaIrO3, а также Cs4IrO4, в котором содержатся плоско-квадратные анионы IrO44– (K.Mader, R. Hoppe, Z. Anorg. Allg. Chem., 1992, 604, 30).
При кипячении раствора гексахлороиридата(IV) в концентрированной серной кислоте в присутствии сульфата калия образуются кристаллы K10[Ir3O(η2-SO4)6(SO4)3], а в присутствии сульфата аммония - K10[Ir3O(η2-SO4)6(SO4)3]. Оба вещества имеют структуру оксиацетов металлов(III) с мостиковыми η2-сульфатными группами. В основе их структур лежит равносторонний треугольник из атомов иридия, в центре которого находится атом кислорода или азота. В оксо-комплексе три дополнительные сульфатные группы являются монодентатными, занимая места, занятые в оксиацетатах молекулами воды. В нем два атома иридия находятся в степени окисления +3, а один – в +4. В нитридном комплексе –два атома иридия(IV) и один иридия(III).
Соединения иридия(V) немногочисленны. Среди них следует упомянуть фосфиновые гидридные комплексы IrH5(PR3)2 (Рис.6.45 (а); Рис.6.45 Строение соединений иридия(V): (а) IrH5(PR3)2, (б) KIrO3), гексафтороиридат, образующийся при фторировании трибромида трифторидом брома в присутствии соли щелочного металла
IrBr3 + CsBr + 2BrF3 ¾¾® CsIrF6 + 3Br2
и иридаты(V), например, KIrO3, образующийся при спекании иридия с надпероксидом калия. Структура этого соединения построена из октаэдров IrO6, соединенных общими вершинами (Hoppe R., Claes K., Journ. of Less-Common Metals, 1975, 43, 129) (Рис.6.45 б).
Дата добавления: 2016-01-03; просмотров: 8481;