Гидроксиды, соли и комплексные соединения элементов 8 группы
При движении вниз по группе возрастает устойчивость высоких степеней окисления. Высшая степень окисления, существование которой у железа однозначно не доказано, легко достижима для рутения и особенно для осмия, который при сгорании на воздухе сразу образует тетраоксид OsO4. В соединениях железо наиболее устойчиво в степенях окисления +2 и +3 (Рис.6.17. Диграмма Пурбе для железа. Пунктиром показана область термодинамической устойчивости воды к окислению и восстановлению, двойным пунктиром – облатсь кинетической устойчивости), рутений – в степени окисления осмий, осмий – в степени окисления +4. Низшие степени окисления представлены преимущественно катионными формами, либо кластерами, наиболее типичными для химии рутения и осмия. В высоких степенях окисления наиболее сильные окислительные свойства характерны для соединений железа, что следует из сопоставления электродных потенциалов ферратов, рутенатов и осматов:
M Fe Ru Os
E°(MO42–/M3+), B pH = 0 2.20 1.56 0.99
Проводя аналогичное сопоставление потенциалов MO4/M0, приведенных в предыдущем разделе и принимая во внимание способность высшего оксида рутения воспламенять органические вещества, можно предположить, насколько сильным окислителем являлся бы тетраоксид железа, если бы его удалось получить.
Низшие степени окисления представлены карбонилами и фосфиновыми комплексами.
При взаимодействии тонкого порошка железа с монооксидом углерода при повышенных температуре и давлении образуется желтая жидкость (т. кип. 103 °C) состава Fe(CO)5, состоящая из тригонально-бипирамидальных молекул (Рис.6.18 а) Рис.6.18 Карбонилы железа: (а) Fe(CO)5, (б) Fe2(CO)9, (в) Fe3(CO)12, (г) [Fe4(CO)13]2–). При облучении ультрафиолетом она превращается в золотисто-желтые кристаллы диядерного карбонила Fe2(CO)9 со связью металл-металл (рис.6.18 б). При нагревании до 100 °C он распадается на Fe(CO)5 и треугольный карбонильный кластер Fe3(CO)12 (рис.6.18 в).
На первый взгляд, кажется неожиданным, что при взаимодействии карбонила Fe(CO)5 с водным или спиртовым раствором щелочи образуется гидридный комплекс:
Fe(CO)5 + 2NaOH = Na[FeH(CO)4] + NaHCO3
Доказано, что на первой стадии гидроксид атакует одну из карбонильных групп, преобразуя ее в карбоксильную, а затем образующаяся железокарбоновая кислота декарбоксилируется (H. Des Abbayes, Journ. Organomet. Chem., 1989, 359, 205):
Fe(CO)5 + OH– ¾® [(OC)4Fe–COOH]–
+OH– +H2O
[(OC)4Fe–COOH]– ¾¾® [(OC)4Fe–COO]– ¾¾® [Fe(CO)4]2– ¾¾® [HFe(CO)4]–
–H2O –CO2 –OH–
Соль Na[HFe(CO)4] является амфотерным электролитом – ее анионы способны как присоединять, так и отщеплять протон:
[HFe(CO)4]–  [Fe(CO)4]2– + H+, K = 2.1×10–13
[Fe(CO)4]2– + H+, K = 2.1×10–13
[HFe(CO)4]– + H+ [H2Fe(CO)4] , K = 1.0×104
Вещество используют в органическом синтезе для гидрирования олефинов.
Взаимодействием пентакарбонила железа(0) с натрием получают тетракарбонилферрат(–2):
Fe(CO)5 + 2Na ¾¾® Na2[Fe(CO)4] + CO.
Это белый порошок, растворимый в воде, но на воздухе быстро окисляющийся. Атом железа находится в центре тетраэдра, образованного четырьмя карбонильными группами. Известны и более сложные кластеры (рис.6.18 г).
Рутений и осмий, наряду с простыми карбонилами M(CO)5, образуют разнообразные карбонильные кластеры, которые подобно высшим боранам подразделяют на клозо-, нидо- и арахно-производные. В состав многих из них входят атомы углерода или азота, а также гидридные лиганды.
Недавно появилось сообщение о синтезе соединения K3FeO2 при выдерживании смеси K6[CdO4], оксида кадмия и железа в течение 40 дней при 450 °C. В структуре этого феррита(I) присутствуют линейные анионы [O–Fe–O]3– (F. Bernhardt, R. Hoppe, Z. Anorg. Allg. Chem., 1993, 619, 969).
ДОПОЛНЕНИЕ. Ферроцен
В 1951 году было обнаружено, что при взаимодействии хлорида железа(II) с циклопентадиеном HCp (см. т.2, с. 40) в присутствии основания образуются желто-оранжевые кристаллы ферроцена – бис(циклопентадиенил)железа(II) Fe(Cp)2. При нагревании в инертной атмосфере вещество возгоняется, благодаря его молекулярному строению. Оно имеет сэндвичевую структуру, то есть напоминает «бутерброд», в центре которого расположен атом железа и сверху и снизу от него циклопентадиенильные кольца, параллельные друг другу, но находящиеся в антипризматической конформации (Рис. 6.19. Молекула ферроцена). Каждый лиганд образует с атомом железа по пять связей при помощи пяти атомов углерода, такой тип координации обозначают η5. Циклопентадиенильные группы надежно защищают атом железа от окисления: вещество оказывается устойчивым не только при хранении на воздухе, но и обработке горячим концентрированным раствором соляной кислоты. При действии азотной кислоты, иода, хлорного железа, пероксида водорода, хлорида меди(II) и других окислителей молекула ферроцена теряет электрон, превращаясь в синий парамагнитный катион ферроцения:
2Fe(Cp)2 + I2 = 2Fe(Cp)2+ + 2I–, E°( Fe(Cp)2+/ Fe(Cp)2) = 0.3 B.
Сильные восстановители разрывают связь Fe-C, восстанавливая ферроцен до железа.
Циклопентадиенильные группы в ферроцене обладают ароматическим характером и, подобно бензолу, способны вступать в реакции электрофильного замещения, например, ацилирования по Фриделю-Крафтсу. Продукты ацилирования диацетилферроцен – рубиново-красные кристаллы, устойчивые на воздухе. Атомы водорода в циклопентадиенильных группах могут быть замещены также на алкильные заместители, сульфо-группы и фосфины.
Некоторые производные ферроцена используют в качестве лекарств при недостатке железа в организме. Известны также рутеноцен и осмоцен.
КОНЕЦ ДОПОЛНЕНИЯ.
Соединения в степени окисления +2.
При растворении железа в разбавленной серной или хлорной кислотах образуются бледно-зеленые растворы, содержащие гексааква-ионы [Fe(H2O)6]2+. В кристаллическом виде эти ионы существуют в некоторых кристаллогидратах, например, соли Мора (NH4)2Fe(SO4)2×6H2O, железном купоросе FeSO4×7H2O, перхлорате Fe(ClO4)2×6H2O. Они представляют собой высокоспиновые октаэдрические комплексы с конфигурацией t2g4eg2:
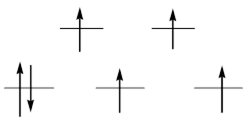
Благодаря различному числу электронов на t2g-орбиталях, ион гексаакважелеза(II) приобретает слабое тетрагональное и ромбическое искажение, особенно заметное в соли Мора, где три независимых расстояния Fe–O составляют 0,185, 0,188 и 0,214 нм. Это является следствием эффекта Яна-Теллера.
Другие соли железа(II) получают восстановлением солей железа(III) железом или по реакциям обмена. Ион [Fe(H2O)6]2+ лишь в слабой степени подвержен гидролизу (Kг,1 = 3,2×10–10), о чем свидетельствует выпадение осадка среднего карбоната при действии соды:
FeSO4 + Na2CO3 = FeCO3¯ + Na2SO4.
Гидроксид железа(II) выделяется в виде белого (крупные кристаллы – светло-зеленые) осадка при добавлении щелочей к растворам солей железа(II). Ввиду легкости окисления препаративный синтез проводят в инертной атмосфере, а исходные растворы готовят с использованием дегазированной воды, не содержащей растворенного кислорода.
Вещество изоструктурно гидроксиду магния и ряда других двухвалентных металлов (кальция, кобальта, никеля, кадмия). Поэтому неудивительно существование минерала амакинита, представляющего собой смешанный гидроксид (Mg, Fe)(OH)2. Все эти вещества имеют слоистую структуру, в которой анионы образуют слегка деформированную гексагональную плотнейшую упаковку, в октаэдрических пустотах которой расположены катионы. Слоистый характер структура приобретает вследствие того, что катионы заполняют пустоты не равномерно, а последовательно занимая каждый второй слой (Рис.6.20 Структурный тип CdI2 (строение гидроксида железа(II); (а) элементарная ячейка, (б) проекция). Это приводит к формированию параллельных друг другу слоев состава МХ2, которые связаны между собой лишь слабыми ван-дер-ваальсовыми силами. Поэтому кристаллы легко раскалываются вдоль плоскости слоев. Данный структурный тип впервые был обнаружен в иодиде кадмия.
Осадок гидроксида железа(II) выпадает в среде, близкой к нейтральной, что свидетельствует о высокой степени его основности. Судя по величине константы (Kb, 2 = 1,3×10–4) он, подобно гидроксиду марганца(II), является более сильным основанием, чем аммиак. При взаимодействии с кислотами гидроксид железа(II) образует соли. Слабая амфотерность этого соединения проявляется лишь в очень крепких щелочных растворах, где при рН >13,5, он начинает медленно растворяться, образуя гидроксокомплексы [Fe(OH)4]2–, [Fe(OH)6]4–.
Все соединения железа(II) являются сильными восстановителями и на воздухе легко окисляются. Белый осадок гидроксида практически мгновенно темнеет, превращаясь в грязно-зеленый осадок, содержащий атомы железа(II) и железа(III) – специалисты, занимающиеся коррозией, называют его «зеленой ржавчиной» и иногда условно записывают в форме гидратированного магнетита Fe3O4×xH2O:
12Fe(OH)2 + 3O2 + ( х – 12)H2O = 4Fe3O4× xH2O
Состав образующегося зеленого осадка в значительной степени зависит от анионов, присутствующих в растворе, так как гидроксид железа(II), окисляясь, захватывает их. При этом слоистая структура гидроксида сохраняется, однако в пустоты между слоями проникают молекулы воды и анионы кислотных остатков, которые компенсируют положительный заряд, вызванный окислением части атомов железа. Состав «зеленой ржавчины» сильно зависит от условий получения, например, из растворов, содержащих хлорид ионы, выделены осадки с различным соотношением железа(II) и железа(III): от Fe32+Fe3+(OH)8Cl×2H2O до Fe2.22+Fe3+(OH)6.4Cl×2H2O, в присутствии сульфат-ионов Fe42+Fe23+(OH)12SO4×nH2O и т.д. (Ph. Refait, S. H. Drissi, et al, Corrosion Science, 1997, 39, 1699 ; 1998, 40, 1547).
При длительном стоянии на воздухе осадок становится бурым, такое изменение окраски соответствует полному окислению железа. Более сильные окислители, например, гипохлорит или бромная вода, сразу позволяют получить гидратированный оксид железа(III):
6Fe(OH)2 + 3Br2 + 6NaOH + ( х – 9)H2O = 2Fe2O3× xH2O¯ + 6NaBr.
В качестве окислителя в щелочной среде можно использовать также и пероксид водорода:
2Fe(OH)2 + H2O2 = Fe(OH)3.
В кислой среде реакция с солью железа(II) носит характер каталитического разложения пероксида.
Фентон в1894 г. обнаружил, что пероксид водорода окисляет винную кислоту и некоторые другие вещества лишь в присутствии ионов железа(II). Механизм реакции на протяжении всего последующего времени являлся предметом дискуссий. Традиционно считается, что окислителем выступает радикал ×OH,образующийся по реакции
Fe2+ + H2O2 ¾® Fe3+ + ×OH + OH–.
Он вступает в реакцию не только с ионами Fe2+, но и с молекулами H2O2, давая радикал ×HO2, который обратно восстанавливает железо(III) до железа(II). Некоторые исследователи предполагают, что окислителем в реакции Фентона служит неустойчивый катион FeO2+, содержащий железо в степени окисления +4. Изучение механизма этой реакции необычайно важно для понимания процессов разложения железосодержащими белками пероксидов, вызывающих старение клетки (H.B. Dunford, Coord. Chem. Rev., 2002, 233-234, 311).
Высокую восстановительную активность гидроксида железа(II) демонстрирует тот факт, что он способен восстанавливать нитрат-ионы до аммиака.
В инертной атмосфере при комнатной температуре гидроксид железа(II) постепенно разлагается на магнетит Fe3O4 и водород, при быстром нагревании до 200 °C обезвоживается, превращаясь в вюстит FeO.
Железо(II) образует соли практически со всеми кислотами, за исключением нитрита, который восстанавливается ионом Fe2+ до NO уже при комнатной температуре. В водном растворе ионы железа(II) окисляются кислородом воздуха, о чем свидетельствует появление желто-коричневой окраски:
4FeSO4 + O2 + 2H2O = 4Fe(OH)SO4.
Крупнокристаллические соли более устойчивы к окислению. Среди них наиболее известна соль Мора (NH4)2Fe(SO4)2×6H2O – двойной сульфат железа(II) аммония со структурой шенита (Сноска: Название вещества связано с именем немецкого химика К. Мора, который впервые использовал его во второй половине XIX века в качестве восстановителя при окислительно-восстановительном титровании). Порошок соли Мора – почти белый, с едва заметным зеленоватым оттенком, а крупные кристаллы имеют красивую бледную зелено-голубую окраску. Они теряют кристаллизационную воду лишь при 100 °C. Высокая устойчивость соли Мора к окислению и выветриванию объясняется развитой системой водородных связей между ионами аммония и молекулами воды, входящими в координационную сферу железа (F.A: Cotton, et al, Inorg. Chem., 1993, 32, 4861) (Рис.6.21. Строение сульфатов железа(II): (а) соли Мора, (б) железного купороса).
Широко употребляемым реактивом служит сульфат железа(II), при комнатной температуре кристаллизующийся из раствора в виде гептагидрата FeSO4×7H2O, называемого железным купоросом. Это вещество образует красивые бледно-голубые кристаллы, которые, в отличие от соли Мора, при хранении выветриваются, превращаясь в белый порошок, а на воздухе постепенно желтеют вследствие окисления. Выветривание железного купороса объясняется тем, что, в отличие от соли Мора, в его структуре [Fe(H2O)6]SO4×H2O присутствует одна молекула внешнесферной воды, которая легко покидает кристаллическую решетку (рис.6.21 б).
В растворе выше 57 °C устойчив тетрагидрат, а выше 64 °C – моногидрат (Рис.6.22 Растворимость сульфата железа(II) в воде), который в инертной атмосфере выдерживает нагревание до 300 °C. Его обезвоживание на воздухе сопровождается частичным гидролизом и окислением до основного сульфата FeOHSO4 и завершается при 700 °C образованием оксида Fe2O3:
100 °C
FeSO4×7H2O ¾¾¾® FeSO4×H2O + 6H2O
150 °C
4FeSO4×H2O + O2 ¾¾¾® 4FeOHSO4
450 °C
6FeOHSO4 ¾¾¾® Fe2(SO4)3 + Fe2O3 + 3H2O
Некоторое количество FeSO4×H2O теряет воду, не успев окислиться, и при 650 °C разлагается на Fe2(SO4)3, Fe2O3 и SO2.
Твердый моногидрат удобно получать дегидратацией купороса в вакууме при 140 °C, выдерживание которого в атмосфере водорода при 300 °C приводит к безводному сульфату железа(II).
Сульфит железа(II) FeSO3×3H2O образуется при насыщении суспензии гидроксида железа(II) сернистым газом. При смешении растворов сульфата железа(II) и сульфита натрия в осадок выпадает двойная основная смешанная соль NaFe2(OH)(SO3)2(H2O).
Нитрат железа(II) Fe(NO3)2×6H2O получают взаимодействием железа с 3 – 5 %-ной азотной кислотой. При слабом нагревании он плавится в кристаллизационной воде, выделяя NO2 и окисляясь до соли железа(III).
Ортофосфат железа(II) встречается в природе в виде минерала вивианита Fe3(PO4)2×8H2O, образующего длинные бесцветные игольчатые кристаллы. На воздухе они становятся ярко-синими, а затем грязно-зелеными вследствие окисления. Белый порошок синтетического вивианита выпадает в осадок при сливании горячих растворов сульфата железа(II) и гидрофосфата натрия. В присутствии ортофосфорной кислоты может быть выделена кислая соль Fe(H2PO4)2(H2O)2. Известен также гидрофосфат FeHPO4(H2O)2.
Синтетический карбонат железа(II) FeCO3 на влажном воздухе быстро окисляется (природный минерал сидерит FeCO3 вполне устойчив), образуя грязно-зеленый осадки переменного состава, напоминающие «зеленую ржавчину», одно из входящих в них веществ имеет формулу Fe42+Fe23+(OH)12CO3×2H2O. Подобно карбонатам щелочноземельных металлов и марганца(II), осадок FeCO3 растворяется в воде при пропускании углекислого газа из-за образования гидрокарбоната Fe(HCO3)2, существующего лишь в растворе. Ион железа(II) в форме гидрокарбоната вместе с ионами кальция и магния обусловливает временную жесткость природных вод. На воздухе он легко окисляется, превращаясь в гидроксид, чем и обусловлено образование бурого налета в местах выхода на поверхность природных вод, богатых железом. При 200 °C средний карбонат разлагается на оксид FeO и углекислый газ, а при более высокой температуре (400 °C) - на Fe3O4, CO и CO2.
При сплавлении оксида железа(II) с кремнеземом образуются силикаты FeSiO3 и Fe2SiO4. Двойные силикаты железа широко распространены в природе. Примером являются минералы оливин (Mg,Fe)2SiO4 и гранат альмандин Fe3Al2(SiO4)3, имеющие островное строение. Из водных растворов получена основная соль Fe3Si2O5(OH)4×2H2O.
Оксалат железа(II) FeC2O4×2H2O выделяется в виде желтого кристаллического порошка при действии на соль Мора оксалатом натрия или при взаимодействии железа с раствором щавелевой кислоты. При его разложении образуется пирофорный оксид железа(II) – раньше по ошибке это вещество принимали за металл. В избытке оксалата натрия соль растворяется, давая комплекс K2[Fe(C2O4)2], который можно высолить спиртом.
Получены двойные соли железа(II) и железа(III), например, Fe+2Fe+3(SO4)4(H2O)2 (Wildner M., Giester G., Z. Kristallographie, 1991, 196, 269), FeFeOPO4, Fe+3Fe5+2 (OH)5(PO4)4(H2O)6.
Соли железа(II) являются сильными восстановителями, хотя и уступают гидроксиду. Они обесцвечивают подкисленный раствор перманганата калия, хлорную и бромную воду, восстанавливают дихромат до солей хрома(III). Для их окисления удобно использовать азотную кислоту:
6FeSO4 + 3H2SO4 + 2HNO3 = 3Fe2(SO4)3 + 2NO + 2H2O.
ДОПОЛНЕНИЕ.
Влияние кислотности среды и комплексообразования на потенциал E(Fe3+/Fe2+).
В стандартных условиях в кислой среде соли железа(III) окисляют иодид-ионы до свободного иода в соответствии со значениями потенциалов:
Fe3+ + e– ® Fe2+, E1° = 0.77 B,
I2 – 2e– ® 2I–, E2° = 0.54 B,
2Fe3+ + 2I– = 2Fe2+ + I2, E1° > E2° (1)
В щелочном растворе реакция (1) протекает в обратном направлении:
2Fe(OH)2 + I2 +2OH– = 2Fe(OH)3 + 2I– (2).
Это объясняется изменением значения потенциала при переходе от солей железа к гидроксидам. В растворе с концентрацией гидроксид-ионов 1 моль/л и при значениях произведений растворимости 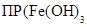 ) = 10–38 и
) = 10–38 и 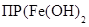 ) = 10–15 получаем
) = 10–15 получаем
E1’(Fe(OH)3/Fe(OH)2) = E1° + 0,059lg 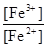 = 0.77 + 0,059lg
= 0.77 + 0,059lg  = –0.59 В ,
= –0.59 В ,
то есть E1’ < E2°.
Иными словами, растворимость гидроксида железа(III) настолько мала, что благодаря удалению ионов Fe3+ из раствора в виде осадка равновесие реакции (1) смещается влево.
Сместить равновесие в сторону окисления иодид-ионов можно и в нейтральной среде, подобрав такую окислительно-восстанoвительную пару, в которой железо(III) образует более устойчивый комплекс, чем железо(II) (Cноска: Kуст [Fe(CN)6]3– = 10–42, Kуст [Fe(CN)6]4– = 10–35). Например, если к раствору, содержащему ионы Fe3+ и I–, добавить цианид, то равновесие также сместится в сторону образования комплекса железа(III):
E1’’([Fe(CN)6]3–/([Fe(CN)6]4–)= E1° + 0,059lg = 0.77 + 0,059lg  = = 0.36 В ,
= = 0.36 В ,
то есть E1’’ < E2°
2[Fe(CN)6]4– + I2 = 2[Fe(CN)6]3–+ 2I–. (3)
Чем ниже потенциал пары Fe(III)/Fe(II), тем легче восстанавливается железо(III) и тем более устойчиво к окислению железо(II). Таким образом, железо в низшей степени окисления необходимо стабилизировать в кислых растворах и в отсутствие цианид-ионов. Все ли лиганды уменьшают устойчивость железа(II) подобно цианиду? Как видно из рис.6.23 (Рис. 6.23. Изменение потенциала пары Fe(III)/Fe(II) в различных комплексных соединениях), аналогично цианиду ведут себя и другие анионные лиганды. Они всегда образуют с ионами Fe3+, имеющими более высокий заряд, более устойчивые комплексы, чем Fe2+. В то же время, железо(II) тяготеет к нейтральным лигандам с развитой π-системой, таким как дипиридил и фенантролин.
КОНЕЦ ДОПОЛНЕНИЯ.
Как правило, железо(II) образует высокоспиновые октаэдрические комплексы, к которым относится рассмотренный выше гексааква-ион. Все они лабильны, однако высокое сродство железа(II) к кислород-донорным лигандам препятствует протеканию реакции замещения воды на аммиак:
[Fe(H2O)6]2+ + 2NH3  [Fe(H2O)4(NH3)2]2+ + 2H2O, K = 160.
[Fe(H2O)4(NH3)2]2+ + 2H2O, K = 160.
Равновесие реакции с этилендиамином, благодаря хелатному эффекту, смещено вправо, и этот комплекс может быть получен из водного раствора:
[Fe(H2O)6]2+ + en [Fe(H2O)4en]2+ + 2H2O, K = 21900.
Синтез аммиакатов проводят при большом избытке аммиака и обязательно в инертной атмосфере, например, насыщая аммиаком раствор, полученный при взаимодействии железа с бромоводородной кислотой:
FeBr2 + 6NH3 = [Fe(NH3)6]Br2
или нагревая порошок железа с хлоридом аммония:
450 °C
2NH4Cl + Fe ¾¾® Fe(NH3)2Cl2 + H2
Водой эти комплексы разлагаются.
Низкоспиновые комплексы железо образует лишь с лигандами сильного поля – цианидом, дипиридилом и фенантролином. Благодаря конфигурации t2g6 с полностью заселенными t2g-орбиталями
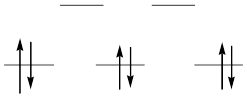
они сочетают высокую термодинамическую и кинетическую устойчивость.
ДОПОЛНЕНИЕ. Кровяные соли.
Гексацианоферрат(II) калия K4[Fe(CN)6] («калий железистосинеродистый») называют желтой кровяной солью. Немецкий фабрикант Дисбах в 1704 г. обнаружил, что при добавлении солей железа к кровяному щелоку – раствору, полученному выщелачиванием сплавленных животных отбросов, поташа и железных опилок, образуется синяя краска, названная берлинской лазурью. В 1752 г Макер обнаружил, что при кипячении лазури с едкой щелочью синяя окраска исчезает и получается желтый раствор, из которого выделяются желтые кристаллы – желтая кровяная соль. В настоящее время ее получают из отработанной «массы Ляминга» (смеси железного купороса, гашеной извести и древесных опилок), применяемой для очистки газов на газовых заводах от примеси циановодорода. Отработанная смесь содержит некоторое количество цианида железа. Ее обрабатывают водной суспензией гидроксида кальция, затем образовавшийся гексацианоферрат(II) кальция извлекают водой и обменным взаимодействием с калийной солью переводят в желтую кровяную соль. В другом промышленным методом синтеза используют взаимодействие суспензии сульфида железа(II) с раствором цианида калия.
Из водных растворов желтая кровяная соль выделяется в виде тригидрата, который при 80 °C обезвоживается, а при прокаливании разлагается на цианид калия, карбид железа Fe3C, азот и, возможно, дициан. При нагревании с разбавленной серной кислотой она дает цианистый водород
2K4[Fe(CN)6] + 3H2SO4(50%) = 3K2SO4 + K2FeFe(CN)6 + 6HCN,
а при действии концентрированной серной кислоты – угарный газ:
K4[Fe(CN)6]×3H2O + 3H2SO4(конц) + 3H2O = 2K2SO4 + FeSO4 + 3(NH4)2SO4 + 6CO.
Ион [Fe(CN)6]4– – типичный одноэлектронный восстановитель, даже многостадийные реакции окисления его кислородом, пероксидом водорода, броматами осуществляются в виде серии одноэлектронных переносов. Например, при действии желтой кровяной солью на раствор бромата калия в слабокислой среде
6[Fe(CN)6]4– + BrO3– + 6H+ = 6[Fe(CN)6]4– + Br– + 3H2O
бромат последовательно восстанавливается до бромида: BrO3– ® BrO2× ® BrO2– ® BrO× ® BrO– ® Br× ® Br– (G. Rabai, I.R. Epstein, Inorg. Chem., 1989, 28, 732).
При подкислении раствора желтой кровяной соли концентрированной соляной кислотой образуется гексацианожелезная (железистосинеродистая) кислота Н4[Fe(CN)6], которая выделяется в виде снежно-белого кристаллического порошка при добавлении эфира или ацетона. Это кислота средней силы (K1 = 10–3), хорошо растворимая в воде и спирте. При нагревании до 100 °C разлагается, выделяя цианистый водород.
Водный раствор желтой кровяной соли при окислении хлором или азотной кислотой краснеет из-за образования гексацианоферрата(III) калия K3[Fe(CN)6] (красной кровяной соли, «железосинеродистого калия»), которая при концентрировании раствора выделяется в виде крупных темно-красных кристаллов.
2K4[Fe(CN)6] + Cl2 = 2K3[Fe(CN)6] + 2KCl.
Эта соль гораздо более ядовита, чем желтая, так как ион [Fe(CN)6]3–, несмотря на большую константу устойчивости, менее инертен, чем [Fe(CN)6]4–: длительным кипячением раствора можно добиться замещения одного цианида на молекулу воды. На холоду действием на красную кровяную соль соляной кислоты получают гексацианожелезную(III) (железосинеродистую) кислоту Н3[Fe(CN)6], которую экстрагируют эфиром. Красная кровяная соль является довольно сильным окислителем, способным превращать гидроксид хрома(III) в хромат, а оксид свинца(II) - в оксид свинца(IV). При кипячении крепкого щелочного раствора она, подобно перманганату и рутенату, выделяет из воды кислород:
t°
4K3[Fe(CN)6] + 4KOH = 4K4[Fe(CN)6] + 2H2O + O2.
Если прокипятить желтую кровяную соль с азотной кислотой, а затем к остывшему раствору прибавить избыток едкого натра, то образуются темно-красные кристаллы нитропруссида Na2[Fe(CN)5NO]. При кипячении с азотной кислотой гексацианоферрат(II) окисляется до гексацианоферрата(III), который вступает в реакцию замещения с образующимся NO. Нитропруссид диамагнитен, то есть является низкоспиновым комплексом железа(III) с конфигурацией t2g6. В нем наблюдается высокая степень π-связывания за счет взаимодействия t2g-орбиталей железа и молекулярных орбиталей лигандов. При добавлении к нитропруссиду сульфид-ионов возникает красное окрашивание, вызванное присоединением серы к нитрозо-группе с образованием [Fe(CN)5(NOS)]4–. Эту реакцию используют для качественного определения серы. Нитрозильный комплекс [Fe(H2O)5(NO)]2+ описан в томе 2 (стр. 188).
Цианидные лиганды, координированные металлом, способны вступать в реакции алкилирования и алкоксилирования. Так, еще в 1854 г было показано, что при взаимодействии H4[Fe(CN)6] с хлороводородом в этиловом спирте образуется хлоридный комплекс с иминоэфиром муравьиной кислоты [Fe(HC(OEt)=NH)6]Cl2, а нагревание Ag4[Fe(CN)6] с этилиодидом приводит к изонитрильному комплексу [Fe(C=N–Et)4(CN)2] (Ю.Н. Кукушкин, Реакционная способность координационных соединений, М., Химия, 1987).
С катионами многих металлов желтая и красная кровяные соли дают ярко-окрашенные осадки комплексных цианидов. Этим пользуются в качественном анализе, например, для определения ионов железа.
Реактивом на ионы Fe2+ служит красная кровяная соль, а на ионы Fe3+ - желтая. В обоих случаях возникает синее окрашивание, которое в первом случае называли турнбулевой синью, а во втором – берлинской лазурью. В настоящее время доказано, что протекающие при этом процессы приводят к образованию одних и тех же соединений, то есть турнбулева синь и берлинская лазурь идентичны, а кажущееся заметным различие в цветовых оттенках обусловлено различным гидратным составом. Известно также, что в зависимости от условий (концентрация, температура) образуются либо синие осадки гексацианоферрата железа Fe4[Fe(CN)6]3 (нерастворимая форма сини), либо коллоидные растворы гексацианоферрата железа-калия KFeFe(CN)6 (растворимая форма сини) (Crumbliss A.L., Lugg P.S., Patel D.L., et al, Inorg. Chem., 1983, 22, 3541). Протекающие при этом процессы представлены уравнениями:
4K3[Fe(CN)6] + 5FeCl2 = Fe4[Fe(CN)6]3¯ + K2Fe[Fe(CN)6] + 10KCl,
K3[Fe(CN)6] + FeCl2 = K[FeFe(CN)6] + 2KCl,
3K4[Fe(CN)6] + 4FeCl3 = Fe4[Fe(CN)6]3¯ + 12KCl,
K4[Fe(CN)6] + FeCl3 = K[FeFe(CN)6] + 3KCl.
Растворимая форма берлинской лазури, иногда называемая прусской синей, содержит ионы Fe3+ в высокоспиновом состоянии и ионы Fe2+ - в низкоспиновом состоянии, соединенные между собой мостиковыми цианидными группами таким образом, что атом углерода соединен с железом(II), а атом азота – с железом(III). Чередующиеся ионы железа(II) и (III) образуют простую кубическую ячейку, в пустотах которой расположены ионы калия (рис.6.24 (а) Строение KFeFe(CN)6). Соединения такого типа известны и с другими переходными металлами (марганцем, хромом), многие из них (NMe4MnFe(CN)6, NMe4MnCr(CN)6) при низких температурах обладают ферримагнетизмом за счет обмена электронами с участием разрыхляющей рπ*-молекулярной орбитали цианидной группы (K.R. Dunbar, R. A. Heintz, Progr. Inorg. Chem., 1997, 45, 283; В.И. Овчаренко, Р.З. Сагдеев, Успехи химии, 1999, 68, 381) (Рис.6.24 б Перекрывание орбиталей). Они представляют собой новый класс магнитных материалов (J.S: Miller, Inorg. Chem., 2000, 39, 4392). Соотношение двух- и трехвалентного железа в растворимой форме равно 1 : 1. Меняя его путем окисления или восстановления ионов железа, мы изменяем состав вещества и его свойства:

В кристаллической структуре восстановление Fe3+ до Fe2+ будет приводить к потере ионов калия, а окисление – к их добавлению (Рис.6.24 в). Прусскую белую, называемую также эвериттовой солью, получают действием на желтую кровяную соль ионов железа(II). На воздухе она быстро окисляется, становясь сначала зеленой, а потом синей. При сливании растворов красной кровяной соли и соли железа(III) осадка не образуется.
Необходимо отметить, что все вещества, полученные из растворов, обязательно содержат кристаллизационную воду, замещающую некоторые группы [Fe+2(CN)6], то есть входящую в координационную сферу Fe(III) (Herren F., Fischer P., Ludi A., Haelg W., Inorg. Chem., 1980, 19, 956). Интенсивная окраска турнбулевой сини связана с переходом электрона от железа(II) к железу(III) (рис.6.24 б).
Для получения крупных кристаллов вещества в 500 мл 10М HCl растворяют 7,5 ммоль хлорида железа(II) и 2,5 ммоль желтой кровяной соли. Открытый стакан с этим раствором помещают на дно сосуда с водой на 8 недель.
КОНЕЦ ДОПОЛНЕНИЯ
Фенантролиновый комплекс железа(II) называют ферроином. Он образует ярко-красные водные растворы, гораздо более устойчивые к окислению, чем обычные соли железа(II). Бромат калия окисляет его лишь в кислой среде. При этом раствор мгновенно приобретает синий цвет:
6[Fe(phen)3]2+ + BrO3– + 6H+ = [Fe(phen)3]3+ + Br– + 3H2O.
красный синий
При подщелачивании красная окраска комплекса восстанавливается, так как в щелочной среде [Fe(phen)3]3+ окисляет бромид-ионы. На этом основано применение ферроина в качестве индикатора при окислительно-восстановительном титровании. Комплексы железа(II) с хелатирующими лигандами часто бывают полиядерными. Примером служит ацетилацетонат, состоящий из тетрамеров Fe4(acac)8. Каждый атом железа в нем имеет координационное число шесть, участвуя как в образовании связей Fe-O, так и в слабых взаимодействиях с атомами углерода.
Известно несколько тетраэдрических высокоспиновых комплексов железа(II), преимущественно галогенидных и псевдогалогенидных ([FeCl4]2–, [Fe(NCS)4]2–).
Химия рутения(II) и осмия(II) представлена комплексными соединениями, которые все без исключения диамагнитны, то есть имеют низкоспиновую конфигурацию. Преобладают октаэдрические комплексы с координационным числом шесть (t2g6). Розовые растворы, содержащие [Ru(H2O)6]2+, получают восстановлением тетраоксида металлическим свинцом:
RuO4 + Pb + 8H+ + 8H2O = [Pb(H2O)6]2+ + [Ru(H2O)6]2+.
Исходный тетраоксид синтезируют взаимодействием диоксида рутения с периодатом натрия. Ионы свинца осаждают добавлением серной кислоты (P. Bernhard, M. Biner, A. Ludi, Polyhedron, 1990, 9, 1095). Ион гексаакварутения(II) удалось выделить в твердом виде в форме тозилата (п-толуолсульфоната) с выходом 80%. Он способен к реакциям замещения с участием хлорид-ионов, молекул ацетонитрила и диметилсульфоксида (Aebischer N., Laurenczg G., Ludi A., et al, Inorg. Chem., 1993, 32, 2810). В инертной среде действием щелочи может быть осажден коричневый осадок гидроксида, который практически не исследован. Ионы [Ru(H2O)6]2+ являются сильными восстановителями, на воздухе они быстро окисляются до [Ru(H2O)6]3+, цвет раствора при этом становится желтым. По этой причине соли рутения(II) с большинством неорганических кислот, анионы которых способны проявлять окислительные свойства, не получены. Обнаружено, что катион гексаакварутения(II) в водном растворе способен восстанавливать даже перхлорат, один из самых индифферентных ионов.
Интересным примером устойчивой соли рутения(II) является двойной сульфат (NH4)2Ru(SO4)2×6H2O со структурой шенита, аналогичный соли Мора.
Оранжевый хлорид гексаамминрутения(II) [Ru(NH3)6]Cl2 образуется при восстановлении трихлорида рутения цинковой пылью в аммиачном буфере:
2RuCl3 + 12NH3 + Zn = 2[Ru(NH3)6]Cl2 + ZnCl2
Он легко окисляется до [Ru(NH3)6]3+ путем внешнесферного одноэлектронного переноса (E°([Ru(NH3)6]3+/[Ru(NH3)6]2+ = 0.24B). При обработке [Ru(NH3)6]Cl2 концентрированной соляной кислотой образуется ярко-синий раствор, содержащий диядерные кластеры
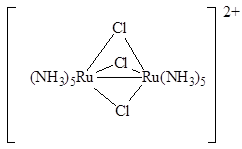
Оба атома рутения в катионе полностью идентичны, поэтому им приписывают дробную степень окисления 2,5. Это вещество предлагалось использовать в качестве краски «рутениевой синей». Яркие «сини» образуются также при восстановлении коммерчески доступного «трихлорида рутения» цинком, амальгамой натрия, другими восстановителями. Они не являются индивидуальными соединениями, а содержат различные комплексы рутения, часто смешанновалентные, например, [Ru5Cl12]–, [Cl3Ru+3(μ-Cl)3Ru+2(μ-Cl)3Ru+3Cl3]4–:
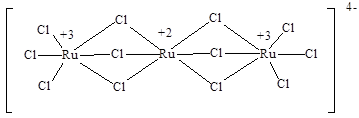
В водном растворе гексааммин [Ru(NH3)6]2+ гидролизуется до пентаммина [Ru(NH3)5(H2O)]2+, в котором молекула воды может быть замещена на другие лиганды. К особенности химии рутения и осмия в низших степенях окисления относится способность образовывать устойчивые комплексы с π-акцепторными лигандами, даже такими, как оксид азота(I) и молекулярный азот (см. том 2, с. 168). Комплекс с азотом может быть получен восстановлением N2O, выступающим в роли лиганда:
[Ru(NH3)5(N2O)]2+ + 2Cr2+ + 2H+ = [Ru(NH3)5N2]2+ + 2Cr3+ + H2O.
Это пример реакции координированных лигандов (том 3, глава 1).
Фотохимическая активация аммиакатов, например, путем облучения их ультрафиолетом, вызывает гидролиз или окисление воды:
hν
[Ru(NH3)5(H2O)]2+ + H+ ¾¾® [Ru(NH3)5(H2O)]3+ + 1/2H2.
В случае лигандов, имеющих сопряженную π-систему, например, ароматическое ядро, фотоактивация приводит к возбуждению, вызванному переходом электрона с t2g-орбитали рутения на π*-орбиталь лиганда. Испуская фотон, комплекс возвращается в основное состояние. На этом основано использование дипиридильного комплекса [Ru(dipy)3]2+, существующего в виде двух оптических изомеров (B. Noble, R.D. Peacock, Inorg. Chem., 1996, 35, 1616) в качестве сенсибилизатора при изучении различных фотохимических процессов, например, радиолиза воды. Диядерные смешанновалентные комплексы рутения(II, III) с пиразином, выступающим в качестве мостикового лиганда служат модельными системами для изучения внутримолекулярного электронного переноса, происходящего с участием ароматической π-системы лиганда (R.J. Crutchley, Adv. Inorg. Chem., 1994, 41, 273;E. Krausz, J. Ferguson, Progr. Inorg. Chem., 1989, 37, 293):

Аналогичные комлпексы с замещенными дипиридилами обладают интенсивной люминесценцией.
Исходным веществом для получения фосфиновых комплексов рутения(II) является трихлорид. При кипячении его в спиртовом растворе с трифенилфосфином образуется RuCl2(PPh3)2, который может быть восстановлен тетрагидроборатом натрия до гидрида RuH2(PPh3)4, подобно пентамминокомплексу, также способного обратимо присоединять молекулярный азот. Благодаря легкости образования гидридов и обратимости взаимодействия, фосфиновые комплексы рутения находят широкое применение в качестве катализаторов гидрирования и гидроформилирования. Комплексы с хиральными фосфинами, такими как BINAP (2,2’-бис(дифенилфосфино)-1,1’-динафтил), играют важную роль в асимметрическом катализе (R. Noyori, Angew. Chem., Int. Ed. Engl., 2002, 41, 2008; W.S. Knowles, Angew. Chem., Int. Ed. Engl., 2002, 41, 1998).
Ацетат рутения(II) Ru2(CH3COO)4L2 представляет собой димер, построенный аналогично ацетату хрома(II). Это соединение отличает способность его поэтапного окисления без изменения строения кластера
+2 – e– +2,+3 – e– +3
Ru2(CH3COO)4L2 ¾¾® [Ru2(CH3COO)4L2]+ ¾¾® [Ru2(CH3COO)4L2]2+
Смешанновалентный комплекс [Ru2(CH3COO)4]Cl образуется в виде коричневого осадка при длительном кипячении трихлорида рутения в уксусной кислоте с добавленным в нее уксусным ангидридом. Изучение его структуры показало, что расстояние Ru=Ru (0,225 нм) соответствует двойной связи металл-металл (A.R. Chakravarty, Proc.Indian Acad. Sci., Sect. A., 1986, 52A, 715), а магнитный момент отвечает наличию трех неспаренных электронов. Это может быть объяснено, если предположить, что π*- и δ*-молекулярные орбитали (см. рис. в главе 4) вырождены.
Химия осмия(II) ограничена в основном соединениями с π-акцепторными лигандами – цианидом, дипиридилом, фосфинами. Некоторые соединения по строению напоминают аналогичные комплексы рутения, но значительно менее устойчивы. Восстановлением катиона пентамминосмия(III) амальгамой цинка в водно-ацетоновом растворе получен катион [Os(NH3)5O=CMe2]2+. Из подобных комплексов могут быть получены соединения молекулярного азота [Os(NH3)5N2]X2. Выпариванием растворов осмата K2OsO6 и цианида калия получены бесцветные кристаллы комплексного цианида K4[Os(CN)6], изоструктурные желтой кровяной соли и тоже химически инертные. Действием на них хлороводорода выделена в кристаллическом виде и сама кислота. Некоторое стабилизирующее действие оказывает на осмий и сульфит-ион, что доказывает возможность получения сульфитного комплекса Na4[Os(SO3)3] и даже вещества OsSO3 неизвестного строения.
Соединения в степени окисления +3.
Данная степень окисления наиболее устойчивая для железа, независимо от кислотности среды. Ионы гексааква-железа(III) входят в состав некоторых кристаллогидратов, однако в растворах в заметной концентрации присутствуют лишь в сильно кислой среде при рН < 1. Имея высокосимметричную конфигурацию t2g3eg2, в которой на каждой из пяти орбиталей находится по одному электрону, он, подобно изоэлектронному ему аква-катиону марганца(II), практически бесцветен, и отличается от него лишь более высокой энергией стабилизации кристаллическим полем, благодаря более высокой степени окисления комплексообразователя. Таким образом, соединения, в состав которых входит ион [Fe(H2O)6]3+ не имеют окраски, например, Fe(ClO4)3×10H2O, Fe(NO3)3×9H2O. В то же время хорошо известно, что растворы, а зачастую и твердые препараты, солей железа(III) окрашены в желтый или коричневый цвет. Это связано с протеканием гидролиза или вхождением аниона соли в координационную сферу железа.
Гидролиз солей железа(III) – сложный и до конца не изученный процесс. На первой стадии происходит депротонирование одной из молекул воды
[Fe(H2O)6]3+  [Fe(OH)(H2O)5]2+ + H+ , K = 1.84×10–3
[Fe(OH)(H2O)5]2+ + H+ , K = 1.84×10–3
Появление гидроксильной группы в координационной сфере атома железа приводит к настолько сильному смещению полосы переноса заряда в сторону больших длин волн, что ее край захватывает видимую область спектра, приводя к появлению типичной для соединений железа(III) желтой окраски.
На второй стадии процесса образовавшиеся гидроксокомплексы объединяются в диамагнитные димеры, содержащие оксо-мостик, который оказывается предпочтительнее двух μ-ОН групп:
2[Fe(OH)(H2O)5]2+ [(H2O)5Fe–O–Fe(OH2)5]4+ + H2O
(Сноска: Недавно такой ион удалось выделить в виде соли [(H2O)5Fe–O–Fe(OH2)5](NO3)4×2(18-crown-6), стабилизированной краун-эфиром, угол Fe–O–Fe составляет 170°: P.C. Junk, B. J. McCool, B. Moubaraki, K.S. Murray, L. Spiccia, Angew. Chem., Int. Ed. Engl., 1999, 38, 2224). На дальнейших стадиях поликонденсации образуются еще более сложные частицы (Сноска: при хранении на воздухе раствора хлорида железа(III) в пиридине были получены кристаллы Hpy[Fe17O16(OH)12(py)12Cl4] (G.W. Powell, H.N. Lancashire, E.K. Brechin et al, Angew. Chem., Int. Ed. Engl., 2004, 43, 5772). В этом соединении центральный атом железа, находящийся внутри тетраэдра из атомов кислорода, соединен кислородными мостиками с двенадцатью атомами железа, находящимися в октаэдрическом окружении, и дополнен еще четырьмя атомами железа, находящимися на внешнем слое полиоксоаниона, которые соединены с атомами хлора. Строние ядра этого «кластера» очень напоминает фрагмент структуры магнетита, поэтому неудивительно, что вещество обладает ферромагнетизмом. Такие молекулярные магниты являются перспективными материалами для хранения информации), а при рН » 2 начинают образовываться коллоидные частицы гидроксида железа(III). Таким образом, многие растворы солей железа(III) при рН > 1 представляют собой коллоидные растворы гидроксида железа(III) – глядя через них на электрическую лампу, можно наблюдать рассеяние света. При усилении гидролиза гидроксид железа(III) выпадает в форме аморфного студенистого желто-коричневого осадка переменного состава Fe2O3×xH2O, который со временем переходит в кристаллическое состояние.
Бурые осадки, образующиеся при действии щелочей и аммиака на водные растворы солей железа(III), также представляют собой гели Fe2O3×xH2O, x = 1 – 5. При старении они переходят в оксогидроксид FeOOH. Истинный гидроксид железа(III) Fe(OH)3 может быть получен сливанием растворов соли железа(III) и аммиака при температуре не выше 2°C, а также при окислении раствора железного купороса дикислородным комплексом кобальта(III) [Co(en)(dien)]2O2(ClO4)4 (S.C:F. Au-Yeung, G. Denes, J.E: Greedan et al, Inorg. Chem., 1984, 23, 1513). Недавно кристаллический гидроксид Fe(OH)3(H2O)1/4 был обнаружен в Австралии в виде минерала, назанного берналитом (Birch W.D., Pring A., Reller A., Schmalle H.W:, American Mineralogist, 1993, 78, 827). Он построен из октаэдров FeO6 соединенных вершинами в трехмерный каркас, в пустотах которого размещены молекулы воды (Рис.6.25 Структура минерала берналита Fe(OH)3(H2O)1/4). При нагревании до 200 °C вещество превращается в оксогидроксид FeOOH, который существует в виде четырех кристаллических модификаций, обозначаемыми первыми четырьмя буквами греческого алфавита. Наиболее устойчив ромбический α-FeOOH (минерал гетит желтого цвета, аналог диаспора AlOOH). Именно он является конечным продуктом старения гелей и золей аморфного гидроксида железа(III). Тетрагональная β-модификация образуется в виде оранжево-желтого осадка при окислении воздухом растворов хлорида железа(II), нагретых до 60 °C (В.П. Чалый, Гидрооксиси металлов, Киев, Наукова Думка, 1972). Коричневый γ-FeOOH (минерал лепидокрокит) изоморфен бемиту, при нагревании в автоклаве до 115 °C переходит в α-форму. Тригональный δ-FeOOH (минерал фероксигит) может быть получен при быстром окислении гидроксида железа(II) пероксидом водорода. В отличие от остальных форм оксогидроксида, он ферромагнитен. Все модификации построены из цепей или слоев октаэров FeO6, соединенных друг с другом общими ребрами (Сноска: строение бемита, изоморфного γ-FeOOH, см. на рис. 4.12(а) в томе 2). Синтетический гетит, используемый в качестве желтого пигмента в производстве красок и эмалей, получают окислением металлического железа, погруженного в раствор соли железа(II), кислородом воздуха или ароматическими нитросоединениями.
Гидроксид железа(III) относится к слабым (Kb ~ 10–11) амфотерным основаниям с преобладающими основными свойствами. Он легко растворим в кислотах с образованием растворов солей железа(III), с концентрированными растворами щелочей при рН > 14 дает гидроксоферраты(III):
t°
3Ba(OH)2 + 2FeOOH + 2H2O = Ba3[Fe(OH)6]2
Ионы [Fe(OH)6]3– существуют только в сильнощелочной среде, а при разбавлении раствора гидролизуются, выделяя оксогидроксид.
Гидротермальный синтез приводит к оксо-гидроксоферратам сложного состава, например, Ba4Fe9O14(OH)6 (Kitahama K., Kiriyama R., Bull. Chem. Soc. Jpn, 1976, 49, 2748), а твердофазный синтез – к ферритам (ферратам(III)), например, BaFeO3, BaFe2O4, BaFe4O7, BaFe12O19. Многие из них являются ферримагнетиками и находят применение в радиоэлектронике и вычислительной технике. Кубические ферриты со структурой шпинели M2+Fe2O4 (M = Co, Ni, Mn, Cu, Mg) представляют собой магнито-мягкие материалы, то есть обладают высокой магнитной проницаемостью, что позволяет им намагничиваться в слабых магнитных полях. Из них изготавливают сердечники трансформаторов, запоминающие устройства, элементы памяти. Гексагональные ферриты («гексаферриты») PbFe12O19 (минерал магнетоплюмбит) и BaFe12O19 являются важными компонентами постоянных магнитов, так как характеризуются высокими значениями намагниченности насыщения, коэрцитивной силы, остаточной намагниченности; они представляют собой магнито-жесткие материалы. Ортоферриты M2+FeO3 имеют структуру перовскита, а ферриты редкоземельных элементов M3+Fe5O12 (M = Y, Sm – Lu) – это синтетические гранаты. Из них наиболее важен железо-иттриевый гранат, ферримагнетик с температурой Кюри 283 °C. Он используется в магнитозапоминающих устройствах, микроволновой и телевизионной аппаратуре. Порошки ферритов получают спеканием смесей оксидов и карбонатов металлов, совместным упариванием растворов солей, соосаждением гидроксидов с последующим их разложением. В технике они используются в основном в виде монокристаллов или пленок (Левин Б.Е., Третьяков Ю.Д., Летюк Л. М., Физико-химические основы получения, свойства и применение ферритов, М., Химия, 1979). Ферриты двух- и трехзарядных катионов термически устойчивы на воздухе, не растворимы в воде, но разлагаются кислотами. Ферриты щелочных металлов (например, Na4Fe12O20, Na3Fe5O9, NaFeO2, Na4Fe2O5, Na14Fe6O16, Na3FeO3, Na8Fe2O7, Na5FeO4) по строению напоминают силикаты – в них присутствуют либо изолированные тетраэдры [FeO4], либо объединенные друг с другом общими вершинами (рис.6.26. Оксоферраты(III) в системе Na/Fe/O).(Sobotka B.M. Moller A. Z. anorg. allg. Chem., 2003, 629, 2063). Эти вещества разлагаются водой до гидратированного оксида, что было положено в основу ферритного способа производства гидроксида натрия, предложенного алхимиками и осуществляемого путем сплавления соды с гематитом и последующего гидролиза феррита:
t°
Na2CO3 + Fe2O3 ¾® 2NaFeO2 + CO2,
NaFeO2 + H2O = NaOH + FeOOH¯.
Гидроксид железа(III) в водных растворах устойчив, как к окислению, так и к восстановлению. Сильные окислители (бром, гипохлорит) способны перевести его в ферраты(VI).
В степени окисления +3 железо образует соли практически со всеми кислотами. Сульфат железа(III) кристаллизуется из водных растворов в виде различных гидратов, содержащих до десяти молекул воды. При осторожном нагревании они дегидратируются, превращаясь в безводную соль Fe2(SO4)3, которая разлагается на гематит и серный ангидрид при 650 °C. Основной сульфат FeOHSO4 рассмотрен при описании железного купороса. Подобно многим другим солям трехзарядных катионов, сульфат железа(III) образует квасцы KFe(SO4)2×12H2O, кристаллизующиеся в форме красивых бледно-фиолетовых октаэдров. Квасцы получают окислением водного раствора сульфата железа(II) азотной кислотой с последующим добавлением сульфата калия. При кипячении раствора они гидролизуются, превращаясь в основные сульфаты KFe3(SO4)2(OH)6 (существует в природе в виде минерала ярозита) и K5Fe3O(SO4)6(H2O)10. Благодаря способности гидролизоваться, сульфат железа(III), наряду с алюмокалиевыми квасцами, используют в качестве флоккулянта при очистке питьевой воды. Образующиеся при его гидролизе хлопья гидроксида адсорбируют многие примеси.
Нитрат железа(III) проще всего получать взаимодействием железа с 50 %-ной азотной кислотой. Соль кристаллизуется в виде лиловых кристаллов, представляющих собой нонагидрат. При слабом нагревании она плавится в своей кристаллизационной воде, а при 125 °C расплав закипает с разложением. Безводный нитрат получают взаимодействием металла с диоксидом азота (Blackwell L.J., Nunn E.K., Wallwork S.C., J. Chem. Soc., Dalton Trans., 1975, 2068). При нагревании смеси Fe(NO3)3×9H2O и CsNO3 на воздухе при 120 °C получены светло-коричневые гигроскопичные кристаллы нитратного комплекса Cs[Fe(NO3)4], в котором атом железа координирован четырьмя бидентатными нитратными группами, то есть имеет координационное число восемь (А.А. Федорова, П.С. Чижов, И.В. Морозов, С.И. Троянов, Журн. неорг. хим., 2002, 47, 2007).
При добавлении к раствору соли железа(III) гидрофосфата натрия выпадает бледно-желтый осадок среднего фосфата FePO4, нерастворимый в уксусной кислоте, но растворимый в соляной, а также в растворе дигидрофосфата. Известно множество кислых и смешанных солей, например, Fe(H3O)(HPO4)2, Fe3(HPO4)4(H2O)4, Fe7(HPO4)(PO4), (H3O)Fe3(HPO4)2(H2PO4)6(H2O)4. При действии на соли железа(III) раствором среднего фосфата натрия образуются основные соли Fe4(PO4)3(OH)3, NaFe3(OH)4(PO4)2, Fe3(OH)3(PO4)2.
Считается, что при взаимодействии солей железа(III) с растворами карбонатов, сульфитов, сульфидов и силикатов происходит выпадение осадка гидроксида вследствие взаимно усиливающегося гидролиза:
Fe2(SO4)3 + 3Na2CO3 + 6H2O = 2Fe(OH)3¯ + 3CO2 + 3Na2SO4.
Карбонат и сульфит железа(III) как средние соли не существуют, однако рыхлая структура образующегося осадка безусловно включает и анионы, содержащиеся в растворе. В избытке сульфит-ионов происходит образование комплексного иона [Fe(SO3)6]9– (Larsson L.O., Niinistoe L., Acta Chem. Scand., 1973, 27, 859). Описана фаза оксокарбоната Fe2O2CO3 (Erdos V., Altorfer, Werkstoffe una Korrosion, 1976, 27, 304). Реакция с силикатом натрия, по-видимому, приводит к образованию гидроксосиликатов, например, Fe2Si4O10(OH)2, встречающегося в природе в виде минерала феррипирофиллита.
При действии на раствор соли железа ацетатом натрия возникает красно-бурое окрашивание, обусловленное вхождением ацетат-ионов в координационную сферу железа. Из таких растворов со временем кристаллизуются оксиацетаты [Fe3O(CH3COO)6(H2O)3]X, где Х – анион, присутствующий в растворе, например, ацетат или хлорид (С.Г. Шоба, И.Г. Кадельник, М. Гданец и др., Журн. структ. хим., 1998, 39, 917). Строение катиона типично для трехвалентных металлов и уже было описано ранее. При кипячении раствора выпадает аморфный бурый осадок оксо-гидроксосоли неизвестного состава.
Взаимодействие ионов железа(III) с сульфид-ионами зависит от кислотности среды. В кислом и нейтральном растворах протекает окислительно-восстановительная реакция, сопровождающаяся помутнением раствора вследствие образования серы. Это, например, имеет место при действии на сульфат железа(III) сульфидом аммония при рН, близком к нейтральному, или сероводородом:
Fe2(SO4)3 + H2S = 2FeSO4 + H2SO4 + S¯.
В щелочных растворах окислительная активность железа(III) понижается, поэтому реакция с сульфидами щелочных металлов, растворы которых имеют сильно щелочную среду вследствие гидролиза, протекает преимущественно с выпадением осадка гидроксида:
Fe2(SO4)3 + 3Na2S + 6H2O = 2Fe(OH)3¯ + 3Na2SO4 + 3H2S
(Сноска: Реакция протекает через стадию образования неустойчивого сульфида железа(III) Fe2S3×nH2O, который может быть осажден в виде черного порошка при температуре ниже 0 °C. На воздухе он разлагается, реагируя с собственной кристаллизационной водой и окисляясь с выделением серы).
Ион Fe3+, таким образом, является слабым окислителем. Он вступает в реакции лишь с сильными восстановителями, такими как сероводород, соли олова(II), гидразин, гидроксиламин, иодид:
4FeCl3 + 2NH3OHCl = 4FeCl2 + N2O + 6HCl + H2O,
2FeCl3 + SO2 + 2H2O = 2FeCl2 + H2SO4 + 2HCl.
Реакции с сероводородом, сернистым газом и иодидом протекают только в кислой среде, так как окислительные свойства катионов переходных металлов усиливаются при понижении рН.
Для железа(III), как и для изоэлектронного ему марганца(II), характерно образование высокоспиновых октаэдрических комплексов, уже обсужденных на примере аква-иона. Замещение молекул воды в координационной сфере железа на гидроксо-группы, как было указано выше, приводит к смещению полосы переноса заряда в видимую область и возникновению желтой окраски. В присутствии роданид-ионов такое замещение сопровожается мгновенным окрашиванием раствора в кроваво-красный цвет:
[Fe(H2O)6]3+ + SCN– = [Fe(H2O)5(SCN)]2+ + H2O , K = 1.1×103
и служит качественной реакцией на ион Fe3+. При достаточном количестве ионов тиоцианата возможно образование как нейтральной соли, выделенной в твердом виде в форме тригидрата Fe(SCN)3×3H2O, так и октаэдрических анионных комплексов вплоть до [Fe(SCN)6]3–. Средняя соль растворима не только в воде, но и в органических растворителях, даже таких малополярных, как эфир. Это позволяет предположить значительную долю ковалентности связи атома металла с кислотным остатком. Строение этого вещества до сих пор неизвестно. Ярко-красные кристаллы K3[Fe(SCN)6] получают растворением оксогидроксида железа в роданистой кислоте в присутствии избытка роданида калия. В водном растворе один из роданид-ионов в координационной сфере железа замещается на молекулу воды.
Аммиакаты железа(III), например, [Fe(NH3)6]Br3, образующийся при взаимодействии твердого безводного бромида железа(III) с газообразным NH3, существуют только в неводных средах, поэтому для осаждения гидроксида железа(III) удобно использовать водный раствор аммиака.
Ацетилацетонат железа(III) Fe(acac)3 выпадает в виде красного осадка при действии на соли железа(III) ацетилацетонатом щелочного металла. Из эфира или бензола он крис
Дата добавления: 2016-01-03; просмотров: 5473;