Порядок и молекулярность реакции.
Число молекул, вступающих в реакцию, определяют молекулярность реакции.
Так, если в реакцию вступает одна молекула, то такая реакция называется молекулярной реакцией. Если в реакции участвуют две молекулы (безразлично, одинаковые или нет), то такая реакция называется бимолекулярной. Встречаются также тримолекулярные реакции.
Реакции более высокой степени молекулярности крайне редки из–за малой вероятности одновременного столкновения большого числа молекул.
Поэтому большинство реакций протекают в несколько элементарных, простых стадий, в которых участвует небольшое число молекул.
Так, например, рассмотренная выше реакция
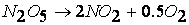
протекает по следующему механизму:
первая стадия
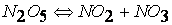
вторая стадия (медленная)
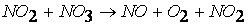
третья стадия
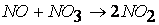
Определить такие стадии – значит определить механизм, или путь реакции.
Скорость всей реакции определяется скоростью её наиболее медленной стадии, которая и определяет механизм.
Поэтому закон действующих масс справедлив только для таких элементарных стадий.
Молекулярность реакции легко определить в случае простых реакций, протекающих в одну стадию. В большинстве же случаев довольно трудно найти молекулярность реакции.
Поэтому вводится понятие порядка реакции, который можно найти из кинетических уравнений, полученных экспериментально.
Порядок реакции по данному веществу равен степени, в которой концентрация данного вещества входит в уравнение скорости реакции.
Сумма показателей степеней, в которых концентрация всех исходных веществ входит уравнение скорости реакции, равна порядку реакции в целом. Порядок химической реакции по веществу совпадает со стехиометрическим коэффициентом реакции лишь в очень простых реакциях, например в реакции синтеза йодистого водорода:
H2 + I2 ® 2HI.
Порядок этой реакции по водороду (первый) и йоду (первый) равны стехиометрическими коэффициентами, а общий порядок реакции (второй) равен сумме стехиометрических коэффициентов в уравнении скорости реакции
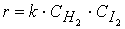
В подавляющем большинстве случаев порядок реакции по веществу отличается от стехиометрических коэффициентов уравнения реакции для этого вещества.
Соответственно общий порядок реакции обычно не равен сумме стехиометрических коэффициентов уравнения реакции.
Например, реакция
NO2 + CO ® CO2 + NO,
при температурах, меньших 298К, протекает по следующему механизму:
первая стадия процесса: NO2 + NO2 ® NO3 + NO
вторая стадия процесса: NO3 + CO ® CO2 + NO2,
причем лимитирующей, т.е. скорость определяющей стадией является первая стадия процесса:
NO2 + NO2 ® NO3 + NO
Тогда, согласно первому постулату химической кинетики, который утверждает, что скорость всей реакции равна скорости его самой медленной стадии, можно записать:
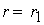 ,
,
где 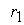 - скорость первой стадии процесса.
- скорость первой стадии процесса.
Согласно второму постулату химической кинетики, который утверждает, что скорость элементарной (одностадийной) реакции пропорциональна концентрации реагирующих веществ в степенях, равных стехиометрическим коэффициентам, получим зависимость скорости реакции
NO2 + CO ® CO2 + NO
от концентрации реагирующих веществ:
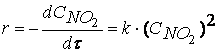
Обратите внимание, что скорость реакции
NO2 + CO ® CO2 + NO
не зависит от концентрации оксида углерода CO.
Уравнение, выражающее зависимость скорости реакции от концентрации каждого вещества, называют кинетическим уравнением реакции в дифференциальной форме.
К сожалению, кинетическое уравнение реакции может быть получено только при её экспериментальном изучении и не может быть выведено из стехиометрического уравнения.
59.Какое явление называется катализом? Отличие гомогенного от гетерогенного. Особенности ферментативного катализа.
Катализ (от греч. κατάλυσις, восходит к καταλύειν — разрушение) — явление изменения скорости химической или биохимической реакции в присутствии веществ, количество и состояние которых в ходе реакции не изменяются (катализаторов).
Термин «катализ» был введён в 1835 году шведским учёным Йёнсом Якобом Берцелиусом.
Для большинства химических процессов немаловажную роль играет скорость их протекания. Одним из широко применяемых методов управления скоростью химической реакции является использование катализаторов. Катализаторами называют вещества, которые изменяют скорость химической реакции, но в результате реакции не расходуются. «Отрицательные катализаторы» — вещества, которые понижают скорость химической реакции, но в процессе реакции не расходуются, — называют ингибиторами. Сам процесс влияния катализатора на скорость химической реакции называют катализом. В зависимости от того, в одинаковых или различных агрегатных состояниях находятся катализатор и реагирующие вещества, различают гомогенный и гетерогенный катализ. При гомогенном катализе катализатор находится в том же агрегатном состоянии, что и реагенты. Если катализатор находится в другом агрегатном состоянии, чем реагенты, то это гетерогенный катализ. Примером гетерогенного катализа является гидрогенизация непредельных жиров на никелевом катализаторе в процессе получения маргарина.
Причины изменения скорости химической реакции в присутствии катализатора обычно связывают с предположением, что в этом случае реакция проходит по принципиально иному механизму, чем в отсутствие катализатора. Для многих реакций экспериментально было подтверждено образование в процессе реакции промежуточных продуктов с участием катализатора.
ФЕРМЕНТАТИВНЫЙКАТАЛИЗ (биокатализ), ускорение биохим. р-ций при участии белковых макромолекул, называемых ферментами (энзимами). Ферментативный катализ- разновидность катализа, хотя термин "ферментация" (брожение)известен с давних времен, когда еще не было понятия хим. катализа.
Первое исследование ферментативного катализа как хим. процесса было выполнено К. Кирхгофом, к-рый в 1814 продемонстрировал фер-ментативную конверсию крахмала в растворимые углеводы.
Заметный вклад в представление о природе ферментативного катализа внесли работы И. Берцелиуса и Э. Мичерлиха, к-рые включили ферментативные р-ции в категорию хим. каталитич. процессов. В кон. 19 в. Э. Фишер высказал гипотезу о специфичности ферментативных р-ций и тесном стерич. соответствии между субстратом иактивным центром фермента. Основы кинетики ферментативных р-ций были заложены в работах Л. Михаэ-лиса (1913).
В 20 в. происходит интенсивное изучение хим. основ ферментативного катализа, получение ферментов в кристаллич. состоянии, изучение структуры белковых молекул и их активных центров, исследование большого числа конкретных ферментативных р-ций и ферментов.
В простейшем случае ур-ние р-ции с участием фермента имеет вид:
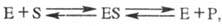
где E - фермент, S - субстрат, ES - фермент-субстратный комплекс (т. наз. комплекс Михаэлиса), P- продукт р-ции.
Превращение субстрата в продукт происходит в комплексе Михаэлиса. Часто субстрат образует ковалентные связи с функц. группами активного центра, в т. ч. и с группами кофермента (см. Коферменты). Большое значение в механизмах ферментативных р-ций имеет основной и кислотный катализ, реализуемый благодаря наличию имидазольных групп остатков гистидина и карбоксильных групп дикарбоно-вых аминокислот.
Важнейшие особенности ферментативного катализа - эффективность, специфичность и чувствительность к регуляторным воздействиям. Ферменты увеличивают скорость хим. превращения субстрата по сравнению с неферментативной р-цией в 109-1012 раз. Столь высокая эффективность обусловлена особенностями строенияактивного центра. Принято считать, что активный центр комплементарен (см. Комплементарность)переходному состоянию субстрата при превращении его в продукт. Благодаря этому стабилизируется переходное состояние и понижается активац. барьер р-ции.
Большинство ферментов обладает высокой субстратной специфичностью, т. е. способностью катализировать превращение только одного или неск. близких по структуре в-в. Специфичность определяется топографией связывающего субстрат участка активного центра.
Активность ферментов регулируется в процессе их биосинтеза (в т.ч. благодаря образованию изоферментов, к-рые катализируют идентичные р-ции, но отличаются строением и каталитич. св-вами), а также условиями среды (рН, т-ра,ионная сила р-ра) и многочисленными ингибиторами и активаторами, присутствующими в организме. Ингибиторамии активаторами могут служить сами субстраты (в определенных концентрациях), продукты р-ции, а также конечные продукты в цепи последоват. превращений в-ва (см. Регуляторы ферментов).
Ферментативные р-ции чувствительны к внеш. условиям, в частности к ионной силе р-ра и рН среды. Влияние т-ры на скорость ферментативной р-ции описывается кривой с максимумом, восходящая ветвь к-рой отражает обычную для хим. р-ций зависимость, выраженную ур-нием Аррениуса. Нисходящая ветвь связана с тепловой денатурациейфермента. Максимум кривой соответствует оптимальной т-ре Tопт, значение к-рой для большинства ферментовлежит в пределах 40-50 0C. Для нек-рых ферментов, особенно ферментов термофильных микроорганизмов, Tопт 80-90 0C. Подробнее о кинетике ферментативных р-ций см. Ферментативных реакций кинетика.
Осн. направления совр. исследований ферментативного катализа- выяснение механизма, обусловливающего высокие скорости процессов, высокую селективность (специфичность действия ферментов), изучение механизмов контроля и регуляции активности ферментов. Оказалось, в частности, что р-ции ферментативного катализа включают большое число стадий с участием лабильных промежут. соед., времена жизни к-рых изменяются в нано- и миллисекундном диапазонах. На активных центрах ферментов протекают быстрые (нелимитирующие) стадии, в результате чего понижается энергетич. барьер для наиб. трудной, лимитирующей стадии.
Установлен механизм регулирования ферментативной активности путем действия ингибитора (или активатора) на специфичный центр белковой молекулы с опосредованной передачей воздействия на активный центр ферментачерез белок. Обнаружены эффекты кооперативного взаимод. неск. молекул субстрата на белковой матрице. Найден способ "жесткого" выведения фермента из процесса посредством индуцированной субстратом необратимой инактивации.
Ферментативный катализ- основа мн. современных хим. технологий, в частности крупномасштабных процессов получения глюкозы и фруктозы, антибиотиков, аминокислот, витаминов и регуляторов, а также тонкого орг. синтеза. Разработаны методы, позволяющие проводить ферментативные р-ции в орг. р-ри-телях, обращенных мицеллах (см.Мицеллообразование). С ферментативным катализом связаны перспективы развития иммуноферментного и биолюминесцентного анализа, применения биосенсоров. Созданы методы, позволившие придать каталитич.активность антителам, обнаружена каталитич. активность у рибонуклеи-новой к-ты (абзимы, рибозимы соотв.).
60.Обратимые и необратимые реакции.
Реакцию называют обратимой, если её направление зависит от концентраций веществ — участников реакции.Например, в случае гетерогенно-каталитической реакции
N2 + 3H2 = 2NH3 (1)
при малой концентрации аммиака в газовой смеси и больших концентрациях азота и водородапроисходит образование аммиака; напротив, при большой концентрации аммиака он разлагается, реакцияидёт в обратном направлении. По завершении обратимой реакции, т. е. при достижении равновесияхимического (См. Равновесие химическое), система содержит как исходные вещества, так и продуктыреакции. Реакцию называют необратимой, если она может происходить только в одном направлении изавершается полным превращением исходных веществ в продукты; пример — разложение взрывчатыхвеществ. Одна и та же реакция в зависимости от условий (от температуры, давления) может бытьсущественно обратима или практически необратима.
Простая (одностадийная) обратимая реакция состоит из двух происходящих одновременноэлементарных реакций, которые отличаются одна от другой лишь направлением химического превращения.Направление доступной непосредственному наблюдению итоговой реакции определяется тем, какая из этихвзаимно-обратных реакций имеет большую скорость. Например, простая реакция
N2O4 ⇔ 2NO2 (2)
складывается из элементарных реакций
N2O4 →2NO2 и 2NO2 →N2O4.
61.Выражение константы равновесия через равновесные концентрации реагирующих веществ.Каков физический смысл константы равновесия.
Количественной характеристикой химического равновесия является константа равновесия, которая может быть выражена через равновесные концентрации Сi, парциальные давления рiили мольные доли Xi реагирующих веществ. Для некоторой реакции
aA + bВ = cC + dD
соответствующие константы равновесия выражаются следующим образом:

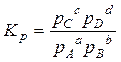
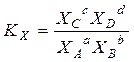
Физический смысл этого принципа заключается в том, что в состоянии термодинамического равновесия сбалансирована каждая реакция в отдельности. Не может быть равновесной ситуации, при которой реакция по одному пути идет преимущественно в одном направлении, а по другому пути – в обратном; компенсация реакции "туда и обратно" имеет место в каждой элементарной стадии.
62.Принцип Ле Шателье.
Принцип Ле Шателье гласит: если система находится в состоянии равновесия, то при действии на нее сил, вызывающих нарушение равновесия, система переходит в такое состояние, в к-ром эффект внеш. воздействия ослабевает. Принцип Ле Шателье определяет смещение хим. и фазовых равновесий при изменении т-ры, давления, состава системы. Напр., повышение т-ры смещает равновесие эндотермич. р-ций в сторону образования конечных продуктов, экзотермич. р-ций - в сторону образования исходных в-в (принцип смещения подвижного равновесия Вант-Гоффа). Повышение давления смещает хим. равновесие в направлении процесса, при к-ром объем системы уменьшается. Введение в систему дополнит. кол-ва к.-л. реагента смещает равновесие в направлении того процесса, при к-ромконцентрация этого в-ва убывает. Сокращенный принцип Ле Шателье - Брауна характеризует смещение равновесияпри участии т. наз. вторичных сил, индуцируемых в системе той силой, к-рая непосредственно воздействует на систему. Если равновесие описывается с помощью обобщенных координат Xi и сопряженных с ними обобщенных сил Уi (i = 1, 2, ..., s, где s - число термодинамич. степеней свободы системы), к-рые изменяются при моновариантном процессе, то интенсивность воздействия силы Yi на координату Xi уменьшается при замене условия постоянства одной из обобщенных сил на условие постоянства сопряженной с этой силой обобщенной координаты. Напр., если для нек-рой системы обобщенными силами являются давление р и т-ра Т, а сопряженные с ними обобщенные координаты - объем V и энтропия S соотв., то неравенство
(дS/дT)p / (дS/дT)V
показывает, что если условие р = const заменить на V = const, значение производной энтропии по т-ре уменьшится. Это приводит к известному соотношению между изобарной и изохорной теплоемкостями: Ср / СV .
Дата добавления: 2015-12-26; просмотров: 1620;