Дифференциальная сканирующая калориметрия (ДСК)
Метод ДТА позволяет получить качественные данные о температурах и знаках фазовых переходов, но этим методом трудно получить количественную информацию об образце или теплоте фазового перехода. Трудности обусловлены тем, что часто неизвестны такие важные факторы, как удельная теплоемкость и теплопроводность образца до и после фазового перехода и другие экспериментальные параметры.
Чтобы получить количественные результаты в качестве камеры для размещения образцов используют дифференциальный калориметр. Держатели анализируемого образца и эталона снабжены собственными электрическими нагревателями. Когда дифференциальная термопара начинает регистрировать напряжение (потенциал), автоматический контролирующий контур подает к более холодному образцу (или эталону) энергию достаточную для компенсации температур между образцом и эталоном. Одновременно с помощью второго электронного контролирующего контура температура образца и эталона линейно увеличивается. Регистрирующее устройство следит за количеством электрической энергии, поданной к образцу или эталону для поддержания изотермических условий. Получаемая термограмма напоминает обычную кривую ДТА, но площадь под пиком в этом случае (ДСК) служит точной мерой энергии, подведенной к анализируемому образцу для компенсации эндотермических эффектов или к эталону для компенсации энергии, излучаемой образцом при возникновении экзотермически эффектов.
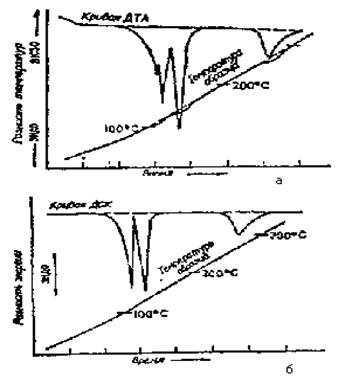
Рис. 23. Разложение Си504*5Н20: а - кривая ДТА б - соответствующая кривая ДСК. Три эндотермы соответствуют потере двух, двух и одной молекул Н20
На рис.23 представлены для сравнения кривые ДТА и ДСК. полученные при термическом разложении СиS0O4*5Н2О. На термограмме ДТА линейный подъем температуры несколько нарушается тепловыми явлениями, происходящими в образце; на термограмме ДСК подобные эффекты невозможны, и пики имеют более правильную форму.
ДСК позволяет проводить точное определение следов примесей в высокочистых органических соединениях по результатам наблюдения за понижением температуры плавления.
ЭЛЕКТРОХИМИЧЕСКИЕ МЕТОДЫ АНАЛИЗА
Краткие теоретические сведения.Потенциометрия – электрохимический метод анализа, заключающийся в измерении электродного потенциала и нахождении связи между его величиной и концентрацией (точнее активностью) потенциалопределяющих компонентов раствора. Используя эту связь, можно определить не только активность и концентрацию ионов, но и другие характеристики: константы диссоциации слабых электролитов, константы устойчивости комплексных соединений, произведения растворимости, стандартные и реальные (формальные) потенциалы окислительно-восстановительных систем, стехиометрические коэффициенты в химических реакциях, число электронов, участвующих в реакциях окисления-восстановления и т.п.
Метод прямой потенциометрии основан на точном измерении величины равновесного электродного потенциала (j) и нахождении по нему активности или концентрации некоторого потенциалопределяющего вещества Х, обычно иона в водном растворе. Поскольку величину потенциала рассчитывают по измеренному значению ЭДС гальванического элемента, включающего индикаторный электрод и электрод сравнения, а потенциал электрода сравнения не зависит от СХ и точно известен, на практике можно ограничиться измерением ЭДС для серии растворов с разными значениями СХ и построением градуировочного графика в координатах ЭДС = f (pCХ). Важнейшим методом прямой потенциометрии (ионометрии) является определение активности ионов водорода и, следовательно, величины pH раствора (логарифм активности ионов водорода, взятый с обратным знаком). Потенциометрическое определение рН является очень точным методом и не требует построения градуировочного графика. При измерении рН можно пользоваться разными индикаторными электродами: водородным, хингидронным, стеклянным, металлоксидными и некоторыми другими.
Стеклянный электрод с Н+-функцией. Стеклянный электрод относится к числу ион-селективных (мембранных) электродов. Установлено, что на поверхности раздела между тонкой мембраной из стекла специального состава и раствором возникает разность потенциалов, величина которой в ряде случаев является функцией ан+ в растворе. С помощью этого электрода можно измерять рН растворов, содержащих сильные окислители и восстановители. Хотя в данном случае механизм возникновения потенциала не является электрохимическим, величина потенциала теоретически соответствует уравнению Нернста:
j = j0 + n lg aН ,
где n = f (T). Точные значения n приведены в справочнике [3], приближенно n = 0,06. У правильно функционирующих стеклянных электродов величины j0 и n в широком диапазоне значений рН сохраняют свое постоянство, то есть зависимость j от рН близка к теоретически рассчитанной прямой вплоть до рН 12. Однако при измерении рН сильнощелочных растворов на потенциал электрода влияют другие катионы, особенно щелочных металлов, что вносит систематическую ошибку. Обычно применяют шарообразные стеклянные электроды, внутреннюю часть которых заполняют раствором HСl, а внешнюю поверхность электрода приводят в соприкосновение с исследуемым раствором. Настройку прибора (градуировку электрода) проводят по буферным растворам с точно известными значениями рН.
Другие ион-селективные электроды. Существуют электроды (преимущественно мембранные), потенциал которых обратимо зависит от активности каких-либо ионов (Х) подобно тому, как потенциал стеклянного электрода зависит от активности ионов Н+. На практике широко используют электроды, селективные к катионам щелочных, щелочноземельных и тяжелых металлов, а также анион-селективные электроды (нитратные, перхлоратные, сульфатные, хлоридные, фторидные и т. п.). Отличие их от рассмотренного выше стеклянного электрода заключается в основном в составе мембраны (стекла, кристаллы солей, вязкие органические жидкости, запрессованные в тонкие полимерные пленки и т.п.).
Работа с ион-селективными электродами аналогична работе со стеклянным электродом с Н+-функцией. Определение рХ проводится по предварительно построенному градуировочному графику, который линеен вплоть до 6-7 единиц рХ. Настройку прибора ведут по растворам с точно известной концентрацией определяемых ионов. Необходимо, чтобы все эти растворы, как и исследуемые растворы с неизвестной концентрацией Х, имели бы одну и ту же температуру, ионную силу и величину рН.
Биамперометрия
Краткие теоретические сведения. Известно, что для прохождения постоянного тока через раствор к электродам надо приложить довольно большое напряжение, обычно для электролиза растворов требуется Е = 1,5 – 2,0 В. Но в некоторых случаях даже при очень небольшом (0,05 – 0,20 В) напряжении, приложенном к паре одинаковых индифферентных электродов, через раствор протекает довольно значительный ток. Этот эффект лежит в основе биамперометрического метода, который в последние годы часто применяется для установления к.т.т. в редоксметрическом титровании. Другое название этого метода – амперометрическое титрование с двумя индикаторными электродами. Метод предложен в 1926 году Фоулком и Боуденом, его отличают необычайная простота аппаратуры, средняя чувствительность (предел обнаружения на уровне 10-5 – 10-6 М) и высокая точность [4; 5]. Схема установки для проведения биамперометрического титрования показана на рис. 1, подобная установка используется и в данной лабораторной работе.
 |
Рис. 1. Схема установки
для биамперометрического титрования:
1- пара одинаковых платиновых электродов; 2- миллиамперметр; 3- потенциометр (реостат); 4 – источник постоянного тока (батарейка); 5 – вольтметр (не обязателен); 6 – бюретка; 7 – магнитная мешалка
Чтобы разобраться в сущности этого метода, рассмотрим процессы, протекающие в разных случаях на электродах, согласно изложению материала Г. Шарло [5]. Следует помнить, что если в каких-либо условиях на границе раздела «электрод-раствор» электрохимическая реакция не идет, сопротивление ячейки становится бесконечно большим и постоянный ток через нее не проходит. Причем необходимо протекание электрохимических процессов и на катоде, и на аноде, то есть на обоих электродах одновременно.
 Предположим, в ячейку залита чистая вода или разбавленный раствор серной кислоты, без примесей окислителей или восстановителей, а затем туда же опущены два платиновых электрода. В этом случае при соответствующей поляризации электродов возможны реакции восстановления воды до водорода (на катоде) и окисления воды до кислорода (на аноде). Судя по известным стандартным потенциалам соответствующих редокс-систем, водород будет выделяться при отрицательном потенциале катода, а кислород – при анодном потенциале, большем чем 1,23 В. Для прохождения тока через раствор и соответственно для процесса электролиза раствора надо на один электрод подать извне потенциал больше 1,23 В, а на другой потенциал меньше 0 В, т. е. напряжение (разность потенциалов), налагаемое на ячейку, должно быть больше 1,23 В. Из-за перенапряжения, связанного с замедленным протеканием электрохимических реакций, на практике приходится давать на ячейку еще большее напряжение (не менее 1,8 В), иначе постоянный ток через раствор почти не идет. Соответствующее напряжение называют напряжением разложения. Данная схема подтверждается соответствующими вольтамперными кривыми (измерениями тока при наложении меняющегося потенциала на каждый из электродов в отдельности) (рис. 2). На вольтамперных кривых принято катодную кривую показывать идущей вниз, анодную – вверх.
Предположим, в ячейку залита чистая вода или разбавленный раствор серной кислоты, без примесей окислителей или восстановителей, а затем туда же опущены два платиновых электрода. В этом случае при соответствующей поляризации электродов возможны реакции восстановления воды до водорода (на катоде) и окисления воды до кислорода (на аноде). Судя по известным стандартным потенциалам соответствующих редокс-систем, водород будет выделяться при отрицательном потенциале катода, а кислород – при анодном потенциале, большем чем 1,23 В. Для прохождения тока через раствор и соответственно для процесса электролиза раствора надо на один электрод подать извне потенциал больше 1,23 В, а на другой потенциал меньше 0 В, т. е. напряжение (разность потенциалов), налагаемое на ячейку, должно быть больше 1,23 В. Из-за перенапряжения, связанного с замедленным протеканием электрохимических реакций, на практике приходится давать на ячейку еще большее напряжение (не менее 1,8 В), иначе постоянный ток через раствор почти не идет. Соответствующее напряжение называют напряжением разложения. Данная схема подтверждается соответствующими вольтамперными кривыми (измерениями тока при наложении меняющегося потенциала на каждый из электродов в отдельности) (рис. 2). На вольтамперных кривых принято катодную кривую показывать идущей вниз, анодную – вверх.
Рис. 2. Вольтамперные кривые электролиза
воды на гладких платиновых электродах:
а – теоретические; б – с учетом перенапряжений из-за замедленного выделения газообразных продуктов
Предположим теперь, что мы ввели в раствор окислитель, например Fe3+. Для этого случая вольтамперные кривые показаны на рис. 3. Напряжение разложения несколько снизится по сравнению с предыдущим случаем, но все же будет более 0,5 В (без учета перенапряжений). При меньших напряжениях ток не пойдет из-за очень большого сопротивления на границе раздела фаз у анода. В данном случае на катоде есть чему восстанавливаться, в растворе имеются ионы Fe3+, но на аноде окисляться нечему, в растворе отсутствует деполяризатор анода. Если залить в ячейку раствор восстановителя FeSO4, то и в этом случае при малых Е ток также не пойдет, но по другой причине – из-за отсутствия деполяризатора катода, слишком большим будет сопротивление на границе раздела фаз у катода.
Если же поместить в ячейку Fe2(SO4)3 и FeSO4 одновременно (вольтамперные кривые показаны на рис.4), то восстановление Fe3+ и окисление Fe2+ на разных электродах идут при очень близких потенциалах, большая разность потенциалов не требуется. Достаточно подать даже малое напряжение (величина Е порядка 0,1 В и менее), чтобы через ячейку пошел ток. Таким образом, для прохождения тока необходимо, чтобы в ячейке присутствовала смесь окисленной и восстановленной форм из одной и той же редокс-системы.

Рис. 3. Вольтамперные кривые электролиза раствора Fe2(SO4)3
Если же поместить в ячейку Fe2(SO4)3 и FeSO4 одновременно (вольтамперные кривые показаны на рис. 4), то восстановление Fe3+ и окисление Fe2+ идут при очень близких потенциалах, большая разность потенциалов не требуется. Достаточно подать даже малое напряжение (величина Е порядка 0,1 В и менее), чтобы через ячейку пошел ток. Таким образом, для прохождения тока необходимо, чтобы в ячейке присутствовала смесь окисленной и восстановленной форм из одной и той же редокс-системы.

Рис.4. Вольтамперные кривые для раствора,
содержащего смесь FeSO4 и Fe2(SO4)3
Однако в некоторых случаях при малых Е ток не идет через раствор даже при одновременном присутствии обоих компонентов редокс пары (например, когда в ячейке находится смесь KMnO4 и MnSO4 или смесь Cr2(SO4)3 и K2Cr2O7). Дело в том, что в этих необратимых системах из-за замедленности катодного и (или) анодного процесса и соответствующих перенапряжений катодная и анодная реакции требуют существенно разных электродных потенциалов. В отличие от обратимой системы Fe3+ – Fe2+, реальная вольтамперная кривая на рис. 5 имеет стелющийся по оси X участок. Необратимыми бывают обычно системы, где кроме перехода электронов в ходе окисления (восстановления) требуется еще разрыв каких-либо межатомных связей, в реакции принимают участие молекулы воды, ионы H+ и др. Разность потенциалов между катодными и анодными участками вольтамперной кривой для раствора, содержащего смесь окисленной и восстановленной формы растворенного вещества, характеризует степень обратимости системы.
|
|

 .
.

| |
|

|
|
|
Рис.5. Вольтамперные кривые для раствора, содержащего смесь Cr2(SO4)3 и K2Cr2O7
(пунктирная кривая – теоретическая, сплошная – эксперимент)
Таким образом, при малых значениях Е протекание тока через биамперометрическую ячейку возможно только при одновременном присутствии в растворе окисленной и восстановленной форм из электрохимически обратимой («быстрой») системы. Ток будет определяться концентрацией той формы, которой в растворе меньше (общую скорость процесса лимитирует либо катодная, либо анодная реакция), а величина избыточной концентрации другой формы практически не влияет на величину тока. Это позволяет определять концентрацию (прямая биамперометрия, биамперометрические датчики). При одной и той же концентрации ток будет тем больше, чем больше приложенное к электродам напряжение E (до определенного предела). При фиксированном значении Е зависимость силы тока от соотношения концентраций двух форм растворенного вещества можно использовать для определения к.т.т. Вид кривых титрования в координатах i – VT весьма разнообразен. Допустим, титруют восстановитель из обратимой системы окислителем, относящимся к необратимой системе. В этом случае кривая титрования имеет максимум (рис. 6).

Рис.6. Кривая биамперометрического титрования ионов Fe2+
бихроматом калия
В начальный момент в растворе имеются только ионы Fe2+, ток близок к нулю, поскольку отсутствует деполяризатор катода. По мере титрования в растворе появляются и накапливаются ионы Fe3+, участвующие в катодной реакции, сопротивление катодной части цепи падает, и ток постепенно возрастает. Если степень оттитрованности равна 50%, концентрации обеих форм равны ( [Fe3+]=[Fe2+] ) и ток максимален. Ближе к концу титрования ток снижается, так как уменьшается концентрация ионов Fe2+ – деполяризатора анода. Сопротивление анодного участка цепи возрастает все больше, и в конечной точке титрования, когда в растворе присутствуют только продукты реакции (Fe3+ и Cr3+), ток близок к 0. При продолжении титрования появляется избыток титранта K2Cr2O7, но одновременное присутствие Cr3+ и Cr2O72- (компоненты необратимой системы) при малых Е не ведет к новому подъему тока, величина i остается близкой к 0.
Если же титровать ионы бихромата раствором соли железа(II), то до точки эквивалентности в растворе будет накапливаться продукт – ионы Fe3+, но ток не пойдет, так как ионов Fe2+ в растворе не будет. Оба компонента обратимой пары появятся только за точкой эквивалентности, соответственно пойдет ток через раствор только после к.т.т. В подобных случаях значения тока (i) в ходе титрования обычно не записывают и кривые не строят, а просто следят за положением стрелки гальванометра. Добавляют титрант до момента резкого броска стрелки, свидетельствующего о к.т.т.
Дата добавления: 2015-10-26; просмотров: 2144;