Система теплорегуляции и теплоотдачи 4 страница
Введение препаратов кальция показано только при подтверждённой гипокальциемии или гиперкалиемии.
Пути введения лекарственных средств при сердечно-лёгочной реанимации могут быть различными. Пока не обеспечен доступ к сосудистому руслу, такие препараты как эпинефрин, атропин, лидока-ин, можно вводить эндотрахеально. Всасывание препаратов из лёгких происходит почти так же быстро, как и при их внутривенном введении.
При осуществлении этой методики необходимо соблюдать следующие правила:
• для лучшей всасываемости лекарственные средства необходимо разводить в достаточном объёме физиологического раствора;
• дозу лекарственного средства необходимо увеличить в 2-3 раза;
• после введения препарата необходимо произвести 5 искусственных вдохов для лучшего распространения лекарственного средства по дыхательным путям.
Показания к внутрисердечному введению лекарственных средств в настоящее время существенно ограничены в связи с вероятностью тяжёлых осложнений (например, гемоперикард, пневмоторакс и др.). Пункция сердца показана только в тех случаях, если ребёнок не интубирован, а доступ к венозному руслу не обеспечен в течение 90 с.
Внутривенный путь введения лекарственных средств наиболее предпочтителен, если у ребёнка уже пунктирована или катетеризирована центральная вена. Попытка обеспечения доступа к центральным, а тем более к периферическим венам во время проведения реанимации крайне затруднена, так как у детей, особенно раннего возраста, стенки сосудов очень эластичны и их просвет поддерживается за счёт АД. Поэтому на фоне остановки кровообращения сосуды спадаются.
Е. Электрокардиография
ЭКГ считают классическим методом мониторирования сердечной деятельности при проведении реанимационных мероприятий. С помощью ЭКГ можно легко диагностировать асистолию, брадикар-дию, фибрилляции и другие нарушения сердечной деятельности. В отдельных случаях прибор может регистрировать практически нормальную электрическую активность сердца при отсутствии сер-
дечного выброса. Такую ситуацию называют электромеханической i диссоциацией. Она может возникнуть при тампонаде сердца, напря-\ жённом пневмотораксе, массивной тромбоэмболии лёгочной арте-|рии, кардиогенном шоке.
IF. Дефибрилляция
\ У детей на фоне фибрилляции желудочков и желудочковой тахи-\ кардии наиболее эффективный метод восстановления кровообраще-[ ния — электрическая дефибрилляция.
I Дефибрилляцию детям проводят сериями из трёх разрядов (2 Дж/кг, !:4 Дж/кг, 4 Дж/кг). Причём если первая серия оказывается неус-> пешной, то на фоне продолжающегося массажа сердца, ИВЛ и медикаментозной терапии необходимо провести вторую серию раз-I рядов.
После успешной реанимации больного следует перевести в специализированное отделение для дальнейшего наблюдения и лечения. :i Показание к прекращению реанимации — отсутствие положитель-[. ного эффекта от проводимых в полном объёме реанимационных мероприятий в течение 30 мин.
\2.6. Организация амбул аторной хирургической помощи. Хирургический ^стационар дневного пребывания
I Оказание высококвалифицированной специализированной по-|мощи в амбулаторных условиях — актуальная задача детской хирур-|;гии. В зависимости от характера заболевания (повреждения) медицинскую помощь оказывают экстренно или в плановом порядке. ^Термин «амбулаторная хирургическая помощь» означает комплекс ^лечебно-диагностических мероприятий, проводимых пациенту в S течение определённого времени (за одно или несколько посещений : детского хирурга), при этом пациента не госпитализируют. Амбулаторную хирургическую помощь оказывают в экстренном или плановом порядке.
Амбулаторную хирургическую помощь можно оказывать в хирур-, гических отделениях, кабинетах поликлиник, травматологических пунктах, приёмных хирургических отделениях, специализированных
центрах амбулаторной хирургии, хирургических стационарах дневного пребывания.
Экстренную хирургическую помощь оказывают немедленно, в момент обращения пациента в лечебное учреждение. Она может быть первым или заключительным этапом лечения — это зависит от характера заболевания (повреждения).
Плановая хирургическая помощь может носить профилактический (например, диспансерный осмотр) или лечебно-диагностический характер. Она включает первичные осмотры, консультации, инвазивные и неинвазивные манипуляции и оперативные вмешательства, проводимые под местным или общим обезболиванием.
Одна из наиболее адекватных форм организации плановой амбулаторной хирургической помощи детям — хирургический стационар дневного пребывания, представляющий собой автономное отделение. В состав хирургического стационара дневного пребывания входят операционные, перевязочные, палаты пред- и послеоперационного пребывания, консультативные кабинеты, служебные и вспомогательные помещения.
Лечение пациента в хирургическом стационаре дневного пребывания начинают с догоспитального этапа, включающего диагностику и отбор пациентов, не имеющих сопутствующих хронических заболеваний, относимых к противопоказаниям к проведению наркоза и оперативного вмешательства в плановом порядке (амбу-латорно). К таким заболеваниям относят пороки развития сердца и магистральных сосудов, отягощенный неврологический анамнез, хронические заболевания почек и др. На этом же этапе проводят инструментально-лабораторное предоперационное обследование. Важный фактор догоспитального этапа — установление психологического контакта хирурга с больным ребёнком и его родителями.
Следующий этап лечения — госпитальный, во время которого проводят операцию (инвазивную манипуляцию). В день операции пациент прибывает в хирургический стационар, где после предоперационного осмотра хирургом и анестезиологом и оформления истории болезни ребёнок остаётся вместе с родителями на несколько часов. За этот период времени проводят оперативное вмешательство и через 3-4 ч после завершения операции пациента выписывают домой.
В хирургическом стационаре дневного пребывания оперируют детей в возрасте старше 1 года с такими заболеваниями, как
паховая и пупочная грыжи, водянка оболочек яичка и семенного канатика, рубцовый фимоз, доброкачественные опухоли мягких тканей и др.
Кроме классических оперативных вмешательств проводят инва-зивные манипуляции с применением низких (криогенных) температур (лечение гемангиом), электрокоагуляции (удаление ангиофибром и других доброкачественных образований), энергии лазеров и сверхвысокочастотного электромагнитного поля (лечение келоидов, не-вусов).
При проведении инвазивных манипуляций и оперативных вмешательств в хирургическом стационаре дневного пребывания широко применяют сочетание местной анестезии (аппликационной, инфиль-трационной и проводниковой) и наркоза; при этом продолжительность последнего не превышает 30—45 мин.
Завершающий этап лечения пациента — постгоспитальный этап, включающий наблюдение в ближайшем послеоперационном периоде до снятия швов или, при необходимости, вплоть до полного выздоровления. В среднем пациент посещает хирурга в послеоперационном периоде 1-2 раза.
Основные преимущества хирургии одного дня:
• значительное уменьшение эмоционально-психической травмы детей, нуждающихся в оперативном лечении;
• снижение до минимума риска внутрибольничного инфицирования;
• наличие алгоритмов отбора пациентов и протоколов оперативного лечения и обезболивания в условиях хирургического стационара дневного пребывания;
• осуществление курации пациента одним хирургом на всех трёх этапах лечения, оптимизация сроков от установки диагноза до оперативного лечения;
• уменьшение объёма догоспитального обследования;
• существенное сокращение затрат на пребывание пациента в стационаре и лечение.
В хирургии одного дня применяют специфические методики, позволяющие уменьшить размеры операционной раны, свести травма-тизацию тканей к минимуму, достичь оптимальных косметического и функционального результатов лечения, сократив время оперативного вмешательства и наркоза.
2.7. Клиническая генетика хирургических болезней у детей
Роль наследственного отягощения в структуре детской заболеваемости и смертности весьма значительна. Так, у 25% пациентов детских клиник диагностируют наследственную и врождённую патологию, а среди умерших детей эта цифра достигает 50%. Наибольшую долю среди таких заболеваний составляют нарушения морфогенеза: врождённые пороки развития, дизрупции, деформации и дисплазии (рис 2-5).
• 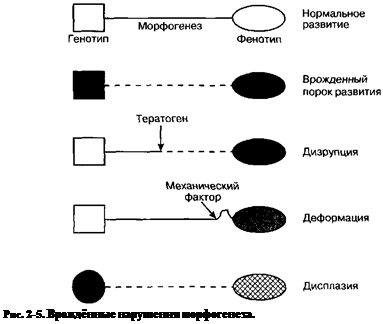
Врождённый порок развития — анатомический дефект органа, возникший в результате первичного, генетически детерминированного нарушения дифференцировки (например, полидактилия, агене-зия или удвоение почки, гипоспадия и т.д.).
• Дизрупция — анатомический дефект органа, возникший в результате вторичного нарушения дифференцировки при нормальном генотипе (например, тератогенные дефекты, вызванные внешними по отношению к эмбриону воздействиями — внутриутробными инфекциями, радиацией, химическими веществами и медицинскими препаратами, заболеваниями беременной).
!• Деформация — аномальная форма или аномальное положение час-' ти тела, вызванное механической причиной в период внутриутробного развития без нарушения дифференцировки (врождённая косолапость, кривошея, врождённая воронкообразная деформация \ грудной клетки и т.д.).
*• Дисплазия — морфологический дефект морфогенеза ткани в результате первичного генетического дефекта (гемангиома, пигментные \ невусы, неоплазии и т.д.).
Вышеперечисленные дефекты развития встречают у новорождённого как единственный признак (частота 3%) и как множественные дефекты (частота 0,7%). У детей с множественными врождёнными дефектами развития чрезвычайно важно диагностировать синдромы (спектр определённых признаков), часто требующие особой терапевтической и хирургической тактики ведения больного.
Синдром
Синдром — состояние, характеризующееся неслучайным сочетанием двух и более врождённых дефектов, вызванных одной причиной. Этой причиной могут быть генная или хромосомная мутация ; (синдром Марфана, синдром Дауна), внутриутробная инфекция (врождённая краснуха), заболевание матери (синдром диабетической эмбриопатии), тератогенное воздействие алкоголя (алкогольный \синдром плода).
):;- В клинической практике диагностика синдромов основана на зна-|нии определённых врождённых дефектов развития, выражающихся 1в фенотипе больного ребёнка. Эти изменения фенотипа или внешнего облика характеризуются комплексом малых аномалий развития | (стигм дизэмбриогенеза), для диагностики и интерпретации которых [•Необходимы определённый опыт и достаточно высокая квалификация врача.
i
Малая аномалия развития
Малая аномалия развития — редкий вариант или врождённое отклонение строения тела — не имеет медицинского значения, т.е. не требует лечения: гипертелоризм (широко расставленные глазные яблоки), преаурикулярные выступы, насечка на мочке ушной раковины, единственная сгибательная складка ладони или мизинца, «шале-видная» мошонка, аплазия ногтя на мизинце (рис. 2-6).
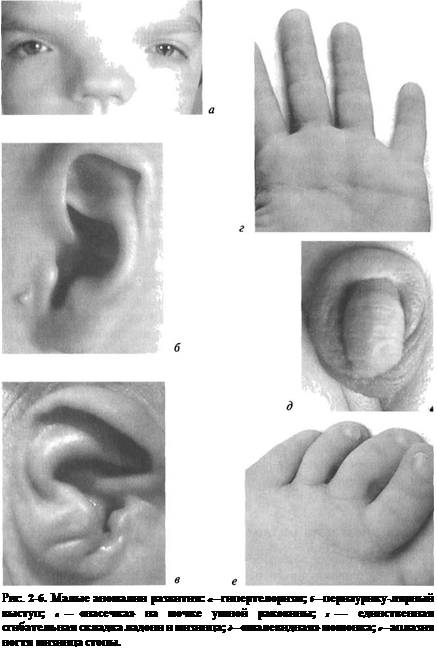
Малые аномалии развития у новорождённых могут быть единичными, или изолированными (частота 14%), а также множественными (две и более малые аномалии развития у ребёнка, частота до 11 %). У новорождённого с наличием трёх и более малых аномалий развития существует 90-процентная вероятность врождённого дефекта развития, в связи с чем необходим поиск такого дефекта. У ребёнка с тремя и более малыми аномалиями развития можно диагностировать определённый синдром с вероятностью 40% — необходима своевременная диагностика. При задержке психомоторного развития и наличии трёх малых аномалий развития в 20% случаев есть вероятность умственной отсталости; большое значение имеет правильный прогноз.
При выявлении у новорождённого трёх и более малых аномалий развития необходимо тщательное УЗИ сердца, головного мозга, почек и органов брюшной полости с целью своевременной диагностики врождённых пороков развития, ещё не имеющих клинических проявлений в этом возрасте. Кроме того, необходима консультация врача-генетика с целью своевременной диагностики определённых синдромов с последующим диспансерным наблюдением.
Классификация OMIM
OMIM (от англ. Online Mendelian Inheritance in Man, менделевское наследование человека в Сети) — постоянно пополняемая и ежедневно обновляемая база данных о генных локусах, фенотипах (включая наследственные заболевания), развиваемая в Университете Джона Хопкинса (США). Представляет совокупность статей. Каждой статье в классификации OMIM присваивается уникальный шестизначный номер, первая цифра которого указывает на способ наследования.
1 —(100000-...) означает аутосомно-доминантный тип наследова-
ния — 9? (статьи, составленные до 15 мая 1994 г., но постоянно обновляемые).
2 — (200000-...) — аутосомно-рецессивное наследование — р (статьи,
составленные до 15 мая 1994 г., но постоянно обновляемые).
3 — (300000-...) — локусы и фенотипы, связанные с Х-хромосомой (К).
4 — (400000-...) — локусы и фенотипы, связанные с Y-хромосомой.
5 — (500000-...) — локусы и фенотипы, связанные с митохондриаль-
ным (цитоплазматическим) наследованием.
6 — (600000-...) — аутосомное наследование (статьи, составленные
после 15 мая 1994 г.).
86 ❖ Хирургические болезни детского возраста ❖ Раздел I
Аллели (аллельные варианты) гена обозначаются шестизначным номером основной статьи (статья гена), за которым следует четырёхзначный номер, обозначающий данный аллель. Например, аллельные варианты (мутации) локуса, кодирующего образование фактора IX свёртывания крови (приводящие к гемофилии В) имеют обозначения от 306900.0001 до 306900.0101 (306900 — обозначение самого локуса).
Звёздочка (*) перед номером локуса или фенотипа означает, что способ наследования для данного локуса или фенотипа доказан (по мнению авторов и редакторов). Отсутствие звёздочки означает, что способ наследования окончательно не установлен.
Символ # перед статьёй означает, что данный фенотип может быть вызван мутацией каким-либо из двух (названных) или более генов.
Классификация OMIM в научном (отчасти клиническом) сообществе стала de facto общепринятой при обозначении и идентификации различных наследственных заболеваний человека.
Синдромология
Примерно у 1 % новорождённых существует неслучайное сочетание нескольких малых аномалий развития и врождённых дефектов. Из них в 40% случаев можно диагностировать тот или иной синдром, а в 60% случаев выделяют так называемые новые синдромы. Это свидетельствует о сложности диагностики синдромов, количество которых в настоящее время приближается к десятку тысяч, причём ежегодно в периодической литературе описывают более сотни новых нозологических форм.
Общая частота большинства синдромальных форм патологии достаточно низка (1 случай на 2000—100000 родов), однако в общей структуре заболеваемости удельный вес синдромальных форм значителен. Так, среди детей с атрезией пищевода частота синдромальных форм патологии достигает 55%, с аноректальными дефектами — 60%, с врождёнными деформациями грудной клетки — 30%.
Отдельные синдромы встречают наиболее часто, поэтому навыками их диагностики должны обладать не только врачи-генетики, но также педиатры и детские хирурги. Например, среди детей с крип-торхизмом и врождёнными пороками сердца встречают синдром Ну-нан, его частота в общей популяции составляет 1 случай на 2000 человек; среди новорождённых с эмбриональной и пупочной грыжей выявляют синдром Беквита—Видеманна с частотой не менее 1 случая на 12000 родов.
Некоторые синдромы хорошо известны в хирургической практике как чрезвычайно важные, способные вызвать серьёзные осложнения. Например, синдром Элерса—Данлоса описан не менее чем в 500 публикациях и нескольких монографиях, так как играет важную роль в хирургии общего профиля, детской и сосудистой хирургии.
Подозрение на синдромальную патологию будет обоснованным у ребёнка с двусторонним поражением, например в случаях двустороннего врождённого дефекта кисти или стопы (полидактилия, врождённая косорукость). Некоторые врождённые пороки развития или малые аномалии развития с высокой вероятностью указывают на синдромальную патологию или определённый врождённый дефект.
Так, преаксиальная полидактилия (удвоение первого пальца кисти или стопы) с большой степенью вероятности свидетельствует о синдромальной патологии, тогда как постаксиальная полидактилия (удвоение мизинца кисти или стопы) обычно бывает изолированным врождённым пороком развития. Наличие полителии (дополнительных сосков или рудиментарных молочных желёз) указывает на высокую вероятность врождённой патологии почек и мочеточников. Врождённое двустороннее отсутствие или гипоплазия первых пальцев кисти свидетельствуют о возможности врождённого дефекта сердца или наличии тромбоцитопатии, что может вызвать серьёзные осложнения во время оперативного вмешательства или в послеоперационном периоде.
Наиболее частыми и важными для хирургов считают заболевания соединительной ткани, биологическую основу которых составляет патология белков внеклеточного матрикса (коллагена, эластина, фиб-.риллина, протеогликанов и гликопротеинов). Эти заболевания представлены наиболее часто встречающимися синдромами (Марфана, Элерса—Данлоса), а также более редкими дисплазиями и мукополи-сахаридозами.
Синдром Марфана
Синдром Марфана (OMIM 154700, дефект гена фибриллина FBN1) — врождённый синдром с аутосомно-доминантным наследованием.
Синдром Марфана следует заподозрить у детей с врождёнными деформациями грудной клетки, аномалиями позвоночника (сколиозом, кифозом), патологической подвижностью суставов и различными
грыжами (паховой, пупочной, диафрагмальной). Очень часто при этом заболевании встречают характерный признак патологии соединительной ткани — пролапс митрального клапана (рис. 2-7).
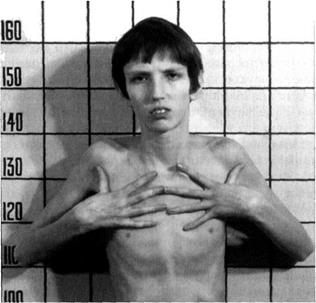
Рис. 2-7. Синдром Марфана.
Диагностические критерии • Скелет
О Главные признаки — килевидная деформация грудной клетки, воронкообразная деформация грудной клетки тяжёлой степени, уменьшение соотношения верхнего/нижнего сегментов тела или увеличение соотношения «размах конечностей/рост» более чем на 1,05, симптом запястья и первого пальца кисти, сколиоз (угол более 20°) или спондилолистез, ограничение разгибания в локтевом суставе (менее 170°), медиальное смещение медиальной лодыжки, плоскостопие, протрузия вертлужной впадины любой степени (по данным рентгенологического исследования).
0 Малые признаки — воронкообразное вдавление грудины, гипермобильность суставов, высокое нёбо с неправильным ростом зубов, черепно-лицевые аномалии (долихоцефалия, гипоплазия
скуловых дуг, энофтальм, ретрогнатия, антимонголоидный разрез глаз).
Скелетную систему считают вовлечённой, если выявляют два главных признака или один главный и два малых признака.
• Глазное яблоко
О Главный признак — эктопия хрусталика.
О Малые признаки — уплощённая роговица (кератометрия), увеличение оси глазного яблока (по данным УЗИ), гипоплазия радужки или гипоплазия цилиарной мышцы.
Глазную систему считают вовлечённой, если выявляют один главный или два малых признака.
• Сердечно-сосудистая система
О Главные признаки — расширение восходящей части аорты с наличием или отсутствием аортальной регургитации и вовлечением синуса Вальсальвы, расслоение стенки восходящей аорты.
О Малые признаки — пролапс митрального клапана, расширение лёгочной артерии при отсутствии клапанного или периферического стеноза, а также если нет видимой причины расширения артерии у лиц старше 40 лет. Кальцификация митрального кольца после 40 лет. Расширение или расслоение нисходящего отдела грудной или брюшной аорты в возрасте старше 50 лет. Систему считают вовлечённой при наличии одного главного или одного малого признака.
• Лёгочная система. Главных признаков нет. Малые признаки — спонтанный пневмоторакс, апикальные псевдокисты лёгкого (по данным рентгенографии). Лёгочную систему считают вовлечённой при наличии одного малого признака.
• Кожа и наружные покровы. Главных признаков нет. Малые признаки — атрофичные стрии, не связанные с изменениями массы тела, беременностью или физическими растяжениями, рецидивирующие грыжи любой локализации. Систему считают вовлечённой при наличии одного главного или одного малого критерия.
• Твёрдая мозговая оболочка. Главный признак — пояснично-крест-цовое расширение (эктазия) эпидурального пространства (по данным КТ спинного мозга).
Условия диагностики синдрома Марфана
• При отсутствии больного родственника (мать, отец, сибс, ребёнок) и негативных ДНК-тестов (ген FBN1) должен присутствовать глав
ный признак в двух различных системах и один малый признак из третьей системы.
• Если выявлена мутация гена FBN1, достаточно одного главного критерия любой системы и вовлечения ещё одной системы. Особенности ведения и осложнения у больных с синдромом Мар-фана:
• затруднение интубации трахеи из-за подвижности височно-ниж-нечелюстного сустава и суставов шейного отдела позвоночника;
• опасность внезапного повышения или снижения АД во время операции;
• осторожное применение мышечных релаксантов при миопати-ческих проявлениях (возможен парадоксальный или пролонгированный эффект);
• возможность летальной желудочковой аритмии и бактериального эндокардита в послеоперационном периоде при пролапсе митрального клапана;
• расширение аорты, образование аневризм и расслоение аорты с возможностью разрыва;
• повышенный риск спонтанного пневмоторакса (4,4%);
• высокая частота пневмоний и хронических эмфиземоподобных изменений;
• снижение жизненной ёмкости лёгких, увеличивающее риск анестезиологических осложнений.
Синдром Элерса-Данлоса
Синдром Элерса-Данлоса (OMIM 130000, 130010, 130020, 130030 и др. в основном мутации генов коллагенов разных типов, а также гена лизил гидроксилазы) — синдром с доминантным или (реже) рецессивным наследованием. Минимальные диагностические критерии: гипермобильность суставов, гиперэластичность кожи с необычной её хрупкостью и необычным заживлением повреждений в виде «папиросной бумаги», участки «шагреневой» или «вельветовой» кожи.
Вероятность синдрома Элерса-Данлоса необходимо учитывать у детей с паховыми и пупочными грыжами, частыми вывихами суставов. Так, при врождённом вывихе тазобедренных суставов у новорождённых это заболевание выявляют в 5% случаев.
В настоящее время выделено не менее 12 типов синдрома Элерса-Данлоса, которые классифицируют с учётом особенностей наи
более тяжёлых клинических проявлений и типа наследования заболевания. Для клиницистов принята практическая классификация синдрома (табл. 2-1).

|- В этой классификации само название типа синдрома даёт ясное Представление о преобладающей картине одного из трёх главных кли-|Ь*ческих проявлений этой патологии: патологической подвижности Йуставов, повышенной эластичности кожи и патологической хрупкости тканей.
I Особенности хирургического ведения и осложнения у больных с Синдромом Элерса-Данлоса:
| • выраженная хрупкость сосудистой стенки (возможность спон-f тайных разрывов крупных артерий, несостоятельность хирургического шва);
• возможность спонтанного разрыва полых органов (кишечника, мочевого пузыря), необходима осторожность при проведении лапароскопии;
• необходима осторожность при проведении ангиографического исследования (возможен разрыв артерий);
• вероятность спонтанного пневмоторакса;
• замедленное формирование послеоперационного рубца (сроки снятия швов увеличены в 1,5—2 раза).
92 О Хирургические болезни детского возраста ❖ Раздел I Синдром Беквита-Видеманна
Синдром Беквита-Видеманна (OMIM 130650, мутация неиденти-фицированного локуса в области pi5.5 хромосомы 11) — синдром со сложным типом наследования. Наиболее характерные проявления при рождении — эмбриональная грыжа, макроглоссия и гигантизм. Минимальные диагностические критерии (рис. 2-8) — большая масса тела при рождении или постнатальное опережение физического развития, дефекты закрытия передней стенки живота (эмбриональная грыжа, пупочная грыжа, диастаз прямых мышц живота), висце-ромегалия (нефромегалия, гепатомегалия, спленомегалия), макроглоссия, характерное лицо (гиперплазия средней трети, гемангиома кожи лба, «насечки» на мочке ушной раковины).
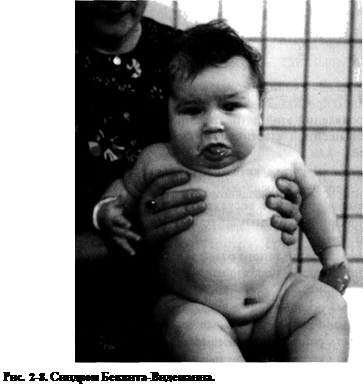
Это заболевание следует заподозрить у детей с эмбриональной или пупочной грыжей, макроглоссией, неонатальной гипоглике
мией и опухолями (нейробластомой, опухолью Вильмса, карциномой печени).
Возможные осложнения у больных с синдромом Беквита—Ви-деманна.
• Вероятность неонатальной гипогликемии (60%) с развитием судорог в послеоперационном периоде.
• Высокая частота (10—40%) эмбриональных опухолей, особенно при наличии нефромегалии или соматической асимметрии тела, что требует наблюдения и проведения УЗИ почек 3 раза в год до 3-летнего возраста, в последующем — 2 раза в год до 14-летнего возраста (своевременная диагностика опухоли Вильмса).
Синдром Нунан
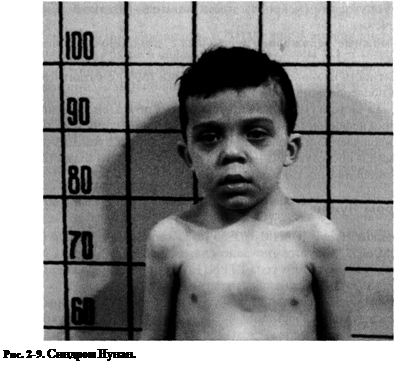
Синдром Нунан (OMIM 163950 [аутосомно-доминантная форма] и OMIM 605275 [аутосомно-рецессивная форма], половина случаев возникает при мутациях гена PTPN11, кодирующего нерецепторную тирозинкиназу). Минимальные диагностические критерии — прена-тально и/или постнатально низкий рост, короткая шея с крыловидными складками, деформация грудной клетки (синостоз рукоятки и тела грудины), необычное лицо (гипертелоризм, лобные бугры, птоз), крипторхизм, врождённый порок сердца — чаще стеноз лёгочной артерии (рис. 2-9).
Особенности ведения и осложнения у хирургических больных с синдромом Нунан:
• развитие тяжёлого хилоторакса/хилоперикарда при повреждении грудного лимфатического протока (кардиохирургия);
• возможна злокачественная гипертермия при проведении анестезии (вероятность 1%);
• высокая вероятность кровотечений в послеоперационном периоде в результате дефицита плазменного фактора свёртывания IX, при болезни фон Виллебранда или нарушении функций тромбоцитов (вероятность до 50%).
Синдром TAR
Синдром TAR (от: thrombocytopenia-absent radius — тромбоцито-пения и отсутствие лучевой кости, OMIM 274000) — синдром с ауто-сомно-рецессивным наследованием. Минимальные диагностические критерии — неонатальная тромбоцитопения, двусторонний врождён
ный дефект кисти, аплазия или гипоплазия лучевой кости с сохранением первого пальца кисти.
Для заболевания характерна морфологическая или функциональная неполноценность мегакариоцитов костного мозга. Для таких больных белки коровьего молока — своеобразный аллерген, вызывающий тяжёлую тромбоцитопению. Хирургические вмешательства также становятся для этих больных стрессовым фактором, вызывающим тромбоцитопению. Кроме основных признаков при этом синдроме часто встречают врождённые пороки почек и сердца.
Особенности ведения больных с синдромом TAR
• специальная диета — исключение коровьего молока с целью профилактики тяжёлой тромбоцитопении;
• до возраста 5 лет противопоказаны плановые хирургические вмешательства (высок риск тромбоцитопении);
• при проведении хирургических вмешательств — переливание свежей, иммунологически совместимой тромбоцитарной массы.
VATER ассоциация
Ассоциация VATER (от: Vertebral defects, Anal atresia, Tracheoesophageal fistula, Esophageal atresia, Radial dysplasy, OMIM 192350) — симптомокомплекс врождённых пороков развития: пороки позвоночника (незарашение дужек, бабочковидные позвонки) — 70%, атре-зия ануса — 80%, трахеоэзофагеальный свищ или атрезия пищевода — 70%, пороки развития почек или радиальные дефекты (аплазия/ гипоплазия лучевых структур кисти) — 65%.
Особенности ведения больных с синдромом VATER: при сочетании у новорождённого атрезии ануса с аномалиями позвоночника или кисти — провести тщательное обследование на наличие трахео-пищеводного свища. При сочетании у новорождённого патологии пищевода и лучевых структур кисти — исключить врождённые пороки почек.
2.8. Малоинвазивные технологии в детской хирургии
Наиболее значимыми событиями последних десятилетий стали бурное развитие и внедрение в широкую клиническую практику эндоскопических методов, коренным образом изменивших лицо современной хирургии.
Общепринятые преимущества эндоскопических технологий — малая травматичность операционного доступа и манипуляций, уменьшение операционной боли, раннее восстановление нарушенных функций организма, уменьшение количества послеоперационных осложнений, сокращение сроков пребывания пациента в стационаре и хороший косметический результат.
Дата добавления: 2015-09-07; просмотров: 2537;