Селективность циклообразования в комплексах переходных металлов
Вспомним, каким трудоемким путем (с общим выходом 0,75%) был впервые получен циклооктатетраен (137,схема 2.65). Этот 10-стадийный синтез был впоследствии воспроизведен другими исследователями всего лишь раз, и на протяжении десятилетий этот углеводород относился к категории экзотических соединений, потенциально интересных по своей химии, но практически не доступных в качестве объекта для исследования.
Ситуация изменилась решающим образом в 1940-х годах, когда Реппе с сотр. [34а] показали, что циклооктатстраен может быть получен с высоким выходом в одну стадию из одного из самых дешевых веществ — из ацетилена. Как это часто бывает, само открытие было сделано в результате случайного наблюдения, но последовавшие вслед за ним углубленные исследования позволили увидеть проявившуюся в нем общую закономерность металлокомп-лексного катализа, ту, которая легла, в частности, в основу нового подхода к построению циклических структур. Рассмотрим этот пример несколько более подробно.
Еще в XIX в. было показано, что ацетилен способен претерпевать термическую олигомеризацию с образованием линейных олигомеров или бензола, но никакого препаративного значения это превращение не могло иметь из-за низкой селективности реакции и малого выхода продуктов. Формально, показанное на схеме 2.135 превращение ацетилена в 137— это реакция точно такого же типа. Что же заставляет эту реакцию протекать селективно как циклогетраме-ризация в присутствии никелевого катализатора (цианида никеля)? Причиной подобной селективности является способность атома никеля образовывать комплекс, в котором четыре молекулы ацетилена расположены так, как это показано в структуре 401.Именно благодаря такой координации и создаются условия, максимально благоприятные для циклоолигомеризации с участием четырех (и только четырех!) молекул ацетилена.
Другими словами, высокоупорядоченное и потому энтропийно крайне невыгодное переходное состояние для образования 137из четырех малых молекул становится достижимым благодаря предорганизции этих молекул как лигандов вокруг центрального атома металла. Интересно, что если проводить эту же реакцию в присутствии трифенилфосфина, способного также служить лигандом, то образуется комплекс типа 402,и конечным продуктом циклоолигомеризации оказывается циклотример, т.е. бензол [34Ь].
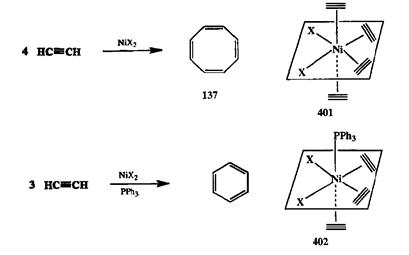 Схема 2.135
Схема 2.135
|
Естественно, что вряд ли оправдано получать сам бензол таким образом, поскольку он в изобилии поставляется другими источниками. Однако сама схема сборки бензольного ядра из трех ацетиленовых единиц (формальная реакция [2+2+2]-циклоприсоединения) оказалась чрезвычайно плодотворной для органического синтеза. Особенно удачными для этой цели явились металлокомплексные катализаторы на основе кобальта и некоторых других переходных металлов.
Детальное исследование хода циклотримеризации ацетиленов в присутствии СрСо(СО)2 показало, что эта реакция начинается с образования комплекса Со — алкин состава 1:1, который далее реагирует со второй молекулой алкина с образованием относительно устойчивого металлоцикла 403.Взаимодействие последнего с третьей молекулой ацетилена, протекает, по-видимому, через промежуточное образование семичленного металлоцикла и приводит к циклотримеру 494(схема 2.136). С препаративной точки зрения особенно удачным явилось применение этой реакции для синтеза различньк полизамещенных производных бензола, таких, как, например, гексаизопро-пилбензол (404а)[34с], которые практически невозможно получить иным способом.
Однако еще более интересные перспективы открылись с разработкой способа соолигомеризации алкинов с 1 ,п-диинами. Как показано на схеме 2.136, подобные реакции могут служить не только общим методом получения разнообразных производных индана и тетралина 405а,B,но и открывают путь к получению таких интересных, но труднодоступных соединений, как бензо-циклобутены [34d] (например, 406).В дальнейшем мы сможем убедиться, насколько разнообразными оказались возможности синтетического использования реакций циклоолигомеризации алкинов, подобные приведенным на схеме 2.136 (см., например, схемы 3.36-3.38 в гл. 3 и схему 4.59 в гл. 4).
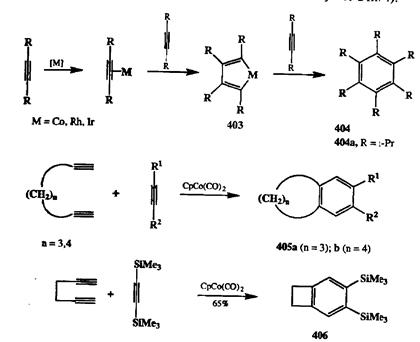 Схема 2.136
Схема 2.136
|
Принцип координации реагентов вокруг центрального атома переходного металла оказался пригодным и для сборки пятичленных циклов по схеме формального [2+2+1]-циклоприсоединения. С точки зрения препаративной значимости наиболее интересной реакцией этого типа является циклоприсоединение аткин+алкен+карбонил (реакция Посона— Кханда), которая в настоящее время широко используется как один из самых удобных методов получения различных соединений, содержащих остаток циклопентенона [34е]. Общая схема реакции показана на примере получения циклопентено-нов простейшего типа 407(см. схему 2.137).
Для полного синтеза полициклических соединений особенно перспективным оказалось использование внутримолекулярного варианта этой реакции, разработанного в группе Шора [34f]. В этом варианте в качестве исходных субстратов используются легко получаемые ji-алкиндикобальтгексакар-бонильные комплексы 1,6- или 1,7-енинов, а результатом превращения является образование бициклического фрагмента, содержащего циклопенте-ноновый остаток (см., например, структуру 408,схема 2.137).
Интересно остановиться на некоторых, сугубо методических вопросах проведения этой реакции. Первоначально циклизацию проводили нагреванием субстрата при довольно высокой температуре (как правило, выше 100°С), из-за чего этот метод оставался малопригодным для термически лабильных полифункциональных соединений. В ходе дальнейших исследований было найдено, что циклоприсоединение резко ускоряется при действии мягких окислителей, например, N-метилморфолиноксида [34g], или при проведении реакции на поверхности силикагеля [34h]. Благодаря этим разработкам стало возможным проводить реакцию в существенно более мягких условиях, что резко расширило границы ее применимости как препаративного метода.
Еще одно преимущество внутримолекулярного варианта реакции Посо-на—Кханда состоит в возможности достаточно быстрой сборки требуемых субстратов с использованием специфических особенностей реакционной способности ц-алкиндикобальтгексакарбонильных комплексов. Как мы уже отмечали ранее (см. разд. 2.2.3.6., схемы 2.43 и 2.44), превращение ацетиленового фрагмента в алкиндикобальтгексакарбонильный комплекс приводит к резкому повышению стабильности соответствующих пропаргильных катионов, и последние могут служить активными электрофилами в реакциях с различными С-нуклеофилами (реакция Николаса, см., например, [18а]). На схеме 2.137 показана схема выполненного Шрайбером [34i] короткого синтеза трициклического соединения 409из ациклического субстрата 410,в которой исчерпывающим образом использованы химические свойства алкиндико-бальтгексакарбонильных комплексов. В самом деле сначала этот фрагмент «работает» на стабилизацию пропаргильного катиона 410а,что позволяет использовать последний для внутримолекулярного пропаргилирования имеющегося в структуре аллилсиланового остатка, приводящего к циклооктино-вому производному 411.На следующей стадии этот же фрагмент выступает в роли реагирующей функции во внутримолекулярной циклизации Посона— Кханда, которая в данном случае протекает с высокой стере оселективностыо. Предорганизция, т.е. предварительное образование жестко фиксированного пространственного расположения лигандов вокруг центрального атома, открывает многочисленные возможности для получения циклических систем с различным размером цикла. Рассмотрим для иллюстрации всего лишь несколько случаев, относящихся к практически важным реакциям.
 Схема 2.137
Схема 2.137
|
Катализируемая комплексами переходных металлов циклоолигемериза-ция 1,3-диенов, таких, как изопрен или бутадиен, была разработана как метод получения восьми- или двенадцатичленных циклических полиенов по схемам [4+4]- или [4+4+4]-циклоприсоединения соответственно [34j]. Показано, что хемо-, регио- и стереоселективность этих превращений зависят от природы используемого металла и могут также регулироваться путем модификации природы дополнительных лигандов или промоторов [34j,k]. Так, бутадиен-1,3 в присутствии комплекса никеля (R3P)2Ni(CO)2 образует ди-мер, *<ис,г<ис-циклооктадиен-1,5 (412,схема 2.138). Из того же субстрата при действии л-комплекса ииклооктен/никель образуется уже тример, транс, /я/»аяс,/ирд«е-циклододекатриен-1,5,9 (413).Изомерный последнему транс, /ярвис,г<ис-циклододекатриен-1,5,9 (414)тоже может быть получен из бутадиена, но на этот раз под действием TiCl4-Et2AlCl.
Катализ комплексами переходных металлов используется также для цик-лообразования в различных вариантах соолигомеризации 1,3-диенов с алке-нами или алюшами [341], а также в синтезе всевозможных гетероциклических сисгем [34т].
Итак, в отличие от других случаев циклоприсоединения (например, таких, как диеновый синтез), в которых изначально задан тип продуктов, которые могут быть получены, реакции с использованием комплексов переходных металлов позволяют получать циклические продукты самых различных типов, причем их природа может варьироваться в очень широких пределах [34п]. Однако следует отметить, что теория катализа комплексами переходных металлов пока развита явно недостаточно и предсказание характера селективности катализируемых ими превращений часто основано скорее на интуиции, чем на строгом знании. Ну что же, честь и слава интуиции, а иногда и простому везению, если с их помощью удается решать сложные научные проблемы!
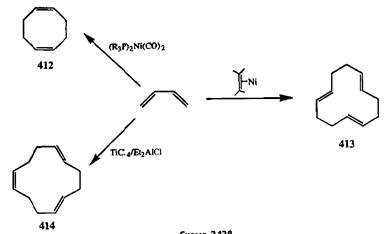 Схема 2.138
Схема 2.138
|
Дата добавления: 2015-04-05; просмотров: 1324;