Радикальные реакции и их роль в синтезе циклических соединений
Как мы уже отмечали, большинство методов образования связей С—С в полном синтезе основано на гетеролитических реакциях или на реакциях циклоприсоединения. Причины того, что гемолитические реакции до недавнего времени находили лишь ограниченное применение в лабораторном органическом синтезе, легко понять, если вспомнить о некоторых известных особенностях механизма этих реакций.
Типичная схема механизма цепной гемолитической реакции включает стадии инициирования (т. е, генерации радикальной частицы), роста цепи и ее обрыва, как это показано в упрощенном виде на схеме 2.139 для случая радикального присоединения по двойной связи.
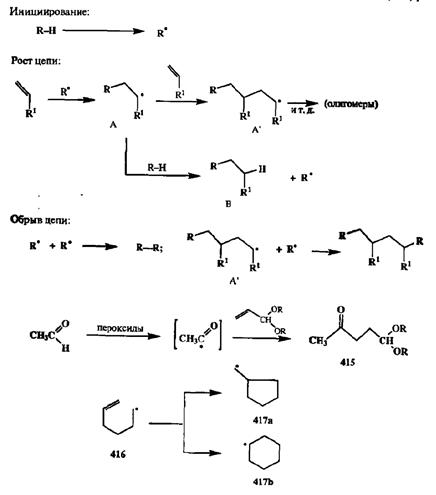 Схема 2.139
Схема 2.139
|
Легко понять, что результат подобной реакции может быть синтетически полезным лишь втом случае, если радикал А, образуемый при присоединении инициатора R' по двойной связи субстрата, может быть немедленно перехвачен с образованием стабильного адцукта. На схеме в роли такого перехватчика показан донор водорода, исходный реагент R—Н. Если подобное не может произойти, то интермедиат А будет реагировать со следующей молекулой исходного алкена, давая интермедиат А, и этот цепной процесс будет продолжаться, приводя в конце концов к образованию смеси олигомсрных продуктов. Поэтому в тех случаях, когда гемолитические реакции присоединения по кратной связи используются в синтезе ациклических продуктов, их обычно проводят в присутствии значительного избытка реагента для обеспечения пре -имущественного образования аддуктов состава 1:1. В качестве типичного примера на схеме 2.139 приведен синтез ацеталя левулинового альдегида (415)реакцией радикального присоединения ацетальдегида (берется в большом избытке) к ацеталю акролеина (гемолитическое гидроацилирование [35а]).
Несмотря на сказанное выше, синтетический потенциал гемолитических реакций огромен, и прежде всего потому, что радикальные частицы относятся к числу высокоактивных интермедиатов, а потому их присоединение по кратным связям протекает достаточно легко и, что немаловажно, в нейтральных условиях. К этому следует также добавить, что подобные реакции малочувствительны по отношению к полярным эффектам в молекуле непредельного субстрата и их можно проводить при наличии самых различных функциональных заместителей, в том числе и таких, присутствие которых исключает саму возможность использования нуклеофильных или электрофильных реагентов.
Осознание этих преимуществ привело к разработке нового подхода к использованию гемолитических реакций, который в настоящее время зарекомендовал себя как один из наиболее эффективных путей синтеза различных циклических соединеиий. Покажем суть этого подхода на модельном примере превращений 5-гексенильного радикала 416.Если генерировать этот радикал из подходящего предшественника (например, гомолизом связи С—I соответствующего 5-гексенилиодида), то немедленным результатом этого будет внутримолекулярная циклизация (ибо, как мы знаем, образование пяти- или шестичленных циклов протекает легче, чем линейная олигомеризация). Образующиеся при этом циклические радикалы 417аили 417Ьне должны быть особенно активными агентами присоединения по кратной связи исходного ковалентного предшественника и при наличии в среде радикальной «ловушки» (восстановителя или окислителя) будут образовывать соответствующие стабильные продукты. Экспериментально было найдено, что основными продуктами реакции исходного 5-гексенилиодида в условиях гемолитических превращений являются производные циклопентана, и, следовательно, циклизация радикального интермедиата 416в основном дает метиленцикло-пентильный радикал (417а)(схема 2.139) [35Ь]. Надо сказать, что преимущественное образование пятичленных циклов при циклизации радикальных интермедиатов — это общая закономерность, не зависящая от конкретных особенностей реакции и природы инициирущих ее реагентов.
Ниже мы рассмотрим более подробно конкретные синтезы с использованием гемолитического присоединения по кратным связям как хорошую иллюстрацию тех новых и уникальных возможностей, которые открываются при вдумчивом использовании вообще-то очень старых реакций.
По-видимому, одним из первых примеров эффективного использования гемолитического присоединения для получения полициклических продуктов был синтез полициклических у-лактонов, разработанный Кори [35с]. Здесь в качестве базовой реакции было выбрано хорошо известное гемолитическое присоединение по кратным связям а-карбонил-радикалов, образующихся при окислении карбоновых кислот солями марганца. Основные стадии этой реакции показаны на схеме 2.140 на примере превращения стирола в лактон 418.
Реакция начинается как присоединение радикала 419(образуемого за счет окисления уксусной хислоты по а-углеродному атому) по двойной связи стирола. Получающийся при этом радикальный интермедиат 419аокисляется в карбокатион 419Ь,который циклизуется с образованием лактона 418.В этом методе используют большой избыток уксусной кислоты и окислителя по отношению к олефину, что практически снимает проблему образования олигомерных продуктов.
Замечательной оказалась эффективность этой реакции во внутримолекулярном варианте, предложенном Кори. Действительно, трансформация субстратов 420—422в соответствующие трициклические лактоны 423—425протекала гладко, в мягких условиях и с высокой регио- и стереоселективностью образования продуктов [35с]. Пожалуй, нелегко было бы предложить другие схемы получения таких лактонов, сравнимые по краткости и простоте исполнения с показанными на схеме 2.140. Отметим также, что синтез требуемых субстратов легко осуществим из доступных исходных с помощью довольно очевидных реакций.
В реакциях, показанных на схеме 2.140, важнейшей стадией является окисление радикального интермедиата в карбокатион как способ обрыва цепи! Альтернативный и, пожалуй, более общий подход был разработан с использованием системы, содержащей и радикальный инициатор, и донор водорода. Чаще всего для этой цели применяется пара: азо-бис-изобутиронит-рил (AIBN) и трибутилстаннан Bu3SnH. В этой системе Bu3SnH играет двойную роль. Первоначально он служит источником радикалов Bu3Sn-, генерируемых под действием AIBN (стадия инициирования, см. схему 2.141),а на завершающей стадии реакции Bu3SnH выступает в роли донора водорода, служащего ловушкой радикалов и агентом роста цепи.
Препаративная ценность этого подхода была убедительно продемонстрирована в работах Сторка [35d], который разработал простой и элегантный синтез систем, содержащих оксациклопентановый остаток, исходя из простейших предшественников. Типичный пример предложенной последовательности превращений показан на схеме 2.141 на примере получения тетра-гидрофуранового производного 426.
 Схема 2.140
Схема 2.140
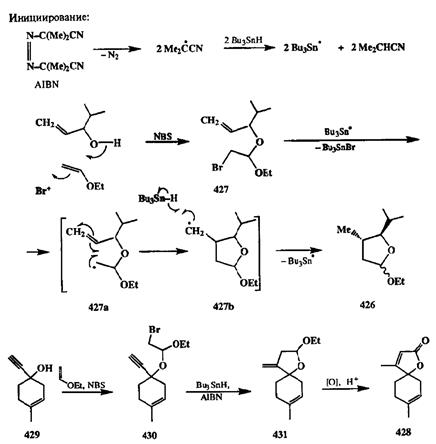 Схема 2.141
Схема 2.141
|
Синтез субстрата циклизации, бромацеталя 427,был выполнен по стандартной схеме сопряженного присоединения по двойной связи, инициируемого электрофильной атакой Вг+ [источник — N-бромсукцинимид (NBS)J и завершающегося присоединением алкокси-нуклеофила (источник — замещенный аллиловый спирт). При взаимодействии 427с системой AIBN-Bu3SnH происходит отрыв атома брома, и образующийся при этом радикальный интермедиат 427ациклизуется с образованием следующего интермедиата 427Ь. Последний отрывает атом водорода от трибутилстаннана, давая продукт 426 и регенерируя при этом радикальную частицу, Bu3Sn-, агент роста цепи. Хорошей иллюстрацией возможностей предложенного метода может служить показанный на схеме 2.141 синтез природного андиролактона (428),исходя из про-паргилового спирта 429и этилвинилого эфира, через стадии образования бромацеталя 430и бициклического продукта 431[35е].
 Схема 2.142
Схема 2.142
|
В развитие этого подхода Сторком [35f] был разработан оригинальный путь получения бициклического соединения 432,ключевого полупродукта в синтезе простагландинов, в котором искусно скомбинирована последовательность внутри- и межмолекулярных реакций радикального присоединения (схема 2.142). В качестве исходного соединения в этом синтезе был взят моноэфир оптически активного диола 433,из которого обычным путем был получен иодацеталь 434.Далее последовала стандартная реакция 434с системой AIBN-Bu3SnH с тем отличием, что в реакционную среду был добавлен избыток енона 435.Благодаря этому последовательность событий — отрыв атома иода с образованием радикала 434аи циклизация последнего с образованием бициклического радикала 434Ь— завершается не отрывом водорода от Bu3SnH, как это обычно бывает (см. схему 2.141), а взаимодействием с акцептором 435,после чего происходит перенос водорода и образуется требуемый продукт 432.Его выход в расчете на исходный 433составил 75%. Всего лишь 4 дополнительных довольно простых стадии потребовалось для превращения 432в простагландин PGF2a.
Дата добавления: 2015-04-05; просмотров: 1266;