Расщепление одинарных связей С-С
Пожалуй, наиболее известный и очевидный пример конструктивной роли «деструктивной» реакции — декарбоксилирование алкилированных производных ацетоуксусного или малонового эфира. По сути дела именно легкость осуществления этой стадии и является предпосылкой для широкого использования всего комплекса синтетических реакций, в которых эти реагенты применяются как синтетические эквиваленты С3- или С2-синтонов.
Разумеется, декарбоксилирование может потребоваться не только в случае синтезов, основанных на применении малонового или ацетоуксусного эфиров. Необходимость в такой операции может возникать достаточно часто, что легко понять, если вспомнить богатые синтетические возможности использования карбоновых кислот и их производных как субстратов в таких реакциях, как диеновый синтез, присоединение по Михаэлю, конденсация Кляйзена, а-алкилирование енолятов и многие другие. Во всех этих случаях непосредственными продуктами реакций являются карбоновые кислоты или их производные. Поэтому универсальность применения подобных методов для синтеза соединений самых различных классов напрямую зависит от возможности достаточно легкого удаления карбоксильной группы из молекулы после того, как ее активирующая роль уже сыграна. Этого удается достичь с помощью многочисленных реакций, химическая сущность которых сводится к разрыву связи R-COOH с отщеплением стабильной молекулы ССЬ или СО. Упомянутое выше декарбоксилирование р-дикарбонильных производных не требует особых ухищрений: оно протекает особенно легко (при умеренном нагревании) благодаря наличию второй карбонильной группы, роль которой ясна из схемы механизма, показанного для модельного примера 436(схема 2.143).
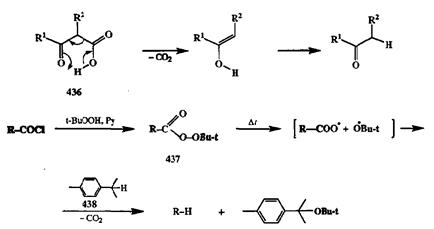 Схема 2.143
Схема 2.143
|
В отсутствие такой активирующей группы приходится применять более жесткие методы удаления «лишней» карбоксильной функции, основанные главным образом на гемолитических реакциях. К таковым относится прежде всего термолиз mpem-бутиловых эфиров пероксикислот 437[36а], которые получают стандартным методом ацилирования т/>ет-бутилгидропероксида хлорангидридом соответствующей карбоновой кислоты. Декарбоксилирование протекает как гемолитический разрыв связи О-О, сопровождающийся элиминированием СО2 и восстановлением образующегося радикала донором водорода, в роли которого обычно используют углеводороды типа 438(см. схему 2.143). Некоторые примеры синтетического использования этого метода будут даны в конце этой главы.
Другим обычно применяемым методом является декарбоксилирование с помощью тетраацетата свинца [36Ь|. В зависимости от строения субстрата, а также условий проведения реакции ее продуктами могут быть либо алкан, либо алкен, либо соответствующее ацетоксипроизводное (схема 2.144). Образованию алканов более всего способствует проведение реакции в присутствии хорошего донора водорода. Часто такое декарбоксилирование проводится в условиях облучения в хлороформе, как это показано на примере превращения циклобутанкарбоновой кислоты в циклобутан [36с]. Напротив, если проводить декарбоксилирование под действием тетраацетата свинца, но в присутствии соокислителя, такого, как ацетат меди, то преимущественным продуктом может стать алкен [36d].
Применение последнего варианта для декарбоксилирования вицинальных дикарбоновых кислот — хороший и общий метод синтеза соответствующих алкенов [36Ь] (схема 2.144). Этот метод часто используется в синтезе напряженных и каркасных углеводородов, где особенно удобно проводить построение углеродного скелета с помощью диенового синтеза с использованием ди-енофилов типа малеиновой кислоты или ацетилендикарбонового эфира [Збе]. Как было неоднократно показано, множество методов образования связи С-С основано на использовании карбонильной группы в качестве активирующей функции, позволяющей вводить самые различные структурные фрагменты по а-атомам углерода, соседним с карбонилом. Продукты таких реакций также являются карбонильными соединениями. Их можно трансформировать далее как без изменения углеродного скелета (например, путем нуклеофильного присоединения по карбонилу), так и с разрывом связи между а-атомом углерода и карбонильной группой. Разрыв связи С-СО достигается с помощью реакции Байера—Виллигсра, состоящей в окислении кетонов надкислотами (или пероксидом водорода в щелочной среде). Эта реакция протекает как нуклеофильное присоединение по карбонильной группе с образованием пероксиадпукта 439,который далее претерпевает перегруппировку (С→O 1,2-сдвиг алкильной группы), образуя сложный эфир (для ацикли-ческих кетонов) или лактон (для циклическихкетонов) (см. схему 2.145) [37а].
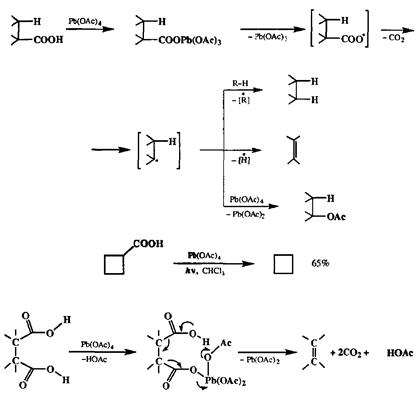 Схема 2.144
Схема 2.144
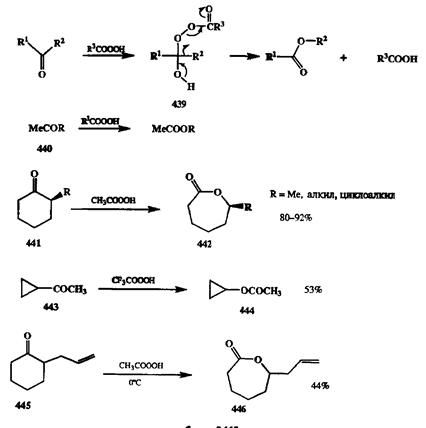 Схема 2.145
Схема 2.145
|
Для этой реакции характерна высокая регио- и стереоселективность. В случае несимметричных кетонов направленность реакции контролируется миграционной способностью алкильной группы, которая в общем коррелирует со стабильностью соответствующих карбокатионов (третичная > вторичная » первичная). Таким образом, с помощью этой реакции любые метилкетоны общей формулы 440могут быть превращены в соответствующие ацетоксипроиз-водные, независимо от характера группы R. Также избирательно проходит окисление а-алкилзамещенных циклогексанонов 441в лактоны 442,производные вторичных спиртов [37Ь]. Возможность проведения окисления в мягких условиях особенно важна, когда речь идет об окислении кислотолабилъ-ных веществ, как, например, в случае реакции 443→ 444[37с]. Интересно, что реакция Байера—Виллигера может протекать селективно даже в тех случаях, когда в молекуле субстрата имеется такая функции, как двойная связь, также способная окисляться надкислотами. В этих случаях требуемая селективность может быть обеспечена просто подбором предельно мягких условий проведения реакции (см. превращение 445→ 446[37d], схема 2.145).
Полное сохранение конфигурации углерода мигрирующей группы также является чрезвычайно важной особенностью окисления по Байеру— Виллиге-ру. Наглядный пример эффективности синтетического применения реакции Байера—Виллигера можно найти в исследованиях Грико [37с] по полному синтезу антибиотика калыдамицина. Одним из ключевых промежуточных продуктов в этом синтезе является лактон 447,в котором содержится три хиралъных центра у соседних атомах углерода (схема 2.146). Исходным соединением для получения этого продукта служил бициклический кетон 448,полученный по схеме диенового синтеза. Благодаря наличию емн-метильной группы в мостике метилирование 448протекало с исключительным образованием энйй-изомера 449.Окисление последнего по Байеру—Виллигеру с последующим омылением лактонного цикла гладко приводило к моноциклической ок-сикислоте 450,в которой уже имеются требуемые хиральные центры. Лакто-низация этой кислоты проходила с аллилъной перегруппировкой и дала бициклический продукт 451,из которого с помощью серии трансформаций был получен циклопентанон 452.Окисление последнего по Байеру—Виллигеру с сохранением конфигурации дало с высоким выходом целевой лактон 447.Последний содержал требуемую комбинацию хиральных центров, причем геометрия этого продукта однозначно определяла стереохимию следующей стадии — метилирования енолята, что позволило превратить 447в стереохимиче-ски чистый лактон 453,из которого далее в несколько стадий был получен один из строительных блоков в синтезе кальцимицина — триол 454.
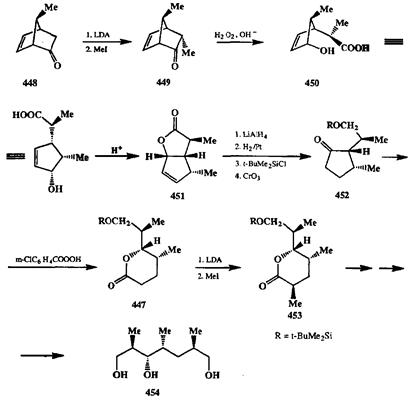 Схема 2.146
Схема 2.146
|
В этой цепочке превращений достаточно сложная задача построения ациклического фрагмента с четырьмя асимметрическими центрами заданной конфигурации бьша решена прежде всего благодаря использованию исходных и промежуточных веществ циклического строения, жесткая геометрия которых обеспечивала надежный контроль стереохимии образования новых центров. Но не менее важным было применение реакции Байера— Виллигера как метода, позволявшего с уверенностью использовать эти преимущества работы с циклическими соединениями, поскольку заранее было известно, что окислительное раскрытие циклов в этих соединениях будет проходить с сохранением конфигурации.
Что касается расщепления неактивированных связей С—С, то здесь препаративное значение имеют лишь методы, основанные на селективном расщеплении 1,2-гликолей под действием периодата (реакция Малачраде, или периодатное окисление) в водной среде или тетраацетата свинца в органических растворителях [38а,B]. Результатом этого превращения является образование двух карбонилсодержаших продуктов. Окисление проходит через образование циклических продуктов, таких, как показанный на схеме 2.147 эфир 455.
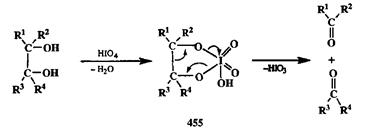 Схема 2.147
Схема 2.147
|
Превращение показанного типа особенно важно в химии полигидрок-сильных соединений, гаких, как углеводы. Многочисленные синтезы хиральных соединений из доступных углеводов основаны на возможности в необходимый момент осуществлять такое расщепление. Рассмотрим, например, как с помощью такой реакции решается задача получения оптически чистого D-глицеринового альдегида (456,схема 2.148).
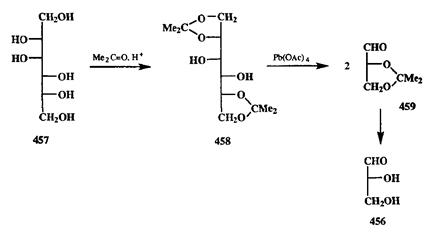 Схема 2.148
Схема 2.148
|
Доступный природный D-маннит (457)сначала переводят в бис-ацето-нид 458,после чего остающуюся 1,2-гликольную группу окисляют тетра-ацетатом свинца. Образующиеся в результате такого разрезания молекулы «половинки» идентичны и представляют собой не что иное, как энантиомерно чистое изопропилиденовое производное D-глицеринового альдегида (459),получаемое таким методом с выходом 70-80% [38с].
Дата добавления: 2015-04-05; просмотров: 1301;