Кислоты, гидроксиды, соли и комплексные соединения. 3 страница
MnO4– + 4H+ + 3e– ® MnO2 + 2H2O, E0 = 1,70 B
TcO4– + 4H+ + 3e– ® TcO2 + 2H2O, E0 = 0,74 B
ReO4– + 4H+ + 3e– ® ReO2 + 2H2O, E0 = 0,51 B
Для химии технеция(IV) и рения(IV) практически не характерно наличие катионных форм, поэтому простые соли этих металлов, несмотря на устойчивость этой степени окисления как к окислению, так и к восстановлению, неизвестны. Большинство изученных соединений представляют собой комплексы, например, [ReCl6]2–, [Tc(NCS)6]2–, Re2Cl4(acac)4 и K4[Re2O2(C2O4)4], в котором два атома металла соединены между собой двумя оксо-мостиками и связью металл-металл (Рис.5.33. Строение аниона в [Re2O2(C2O4)4]4–) (Lis I., Acta Cryst. B., 1975, 31, 1594). Поэтому здесь расстояние Re–Re (0,236 нм) оказывается короче, чем в металлическом рении (0,274 нм). Комплексные соединения рения, как правило, более устойчивы к гидролизу, окислению, разложению, чем аналогичные соединения технеция.
ДОПОЛНЕНИЕ. Дисульфид рения
Дисульфид рения ReS2 представляет собой черный порошок, устойчивый на воздухе, нерастворимый в воде, растворах щелочей, соляной и серной кислот, сульфидов щелочных металлов. Он начинает формироваться из простых веществ уже при комнатной температуре, но реакция завершается при нагревании до 1000 оС. Из других путей синтеза дисульфида можно использовать восстановление высшего сульфида в токе Н2 уже при комнатной температуре, сплавление перрената КReО4 с серой и содой, нагревание оксидов ReО3 или ReО2 с серой или в атмосфере Н2S. В структуре ReS2 атомы расположены между плотно упакованными слоями из атомов серы (Рис.5.34. Строение ReS2). (Murray H.H., Kelty S.P., Chianelli R.R., Day C.S., Inorg.Chem., 1994, 33, 4418) Диcульфид рения устойчив на воздухе без нагревания, выше 300 оС сгорает, выделяя оксиды рения и серы. Его нагревание до 1000 оС без доступа воздуха приводит к образованию металла, содержащего примесь серы. Такой же продукт образуется при восстановлении дисульфида водородом при 700 оС. Горячая азотная кислота окисляет ReS2 до рениевой кислоты. Известен также и дисульфид технеция, полученный из высшего сульфида нагреванием с серой при 1000 оС, или термолизом.
КОНЕЦ ДОПОЛНЕНИЯ
Химия марганца, технеция и рения в степенях окисления +5, +6, +7
Химия кислородных соединений в высших степенях окисления наиболее разнообразна у технеция и рения, а для марганца она представлена тетраоксоманаганат-ионом MnO4n–, свойства которого зависят от степени окисления атома марганца. Манганаты(V), например, K3MnO4 исторически называют гипоманганатами, манганаты(VI) (K2MnO4) – манганатами, а манганаты(VII) (KMnO4) – перманганатами. Согласно диаграмме вольт-эквивалент-степень окисления (рис.5.2) и значениям стандартных электродных потенциалов, перманганаты устойчивы в кислых, нейтральных и слабощелочных средах, а гипоманганаты и манганаты – в сильнощелочных. Это необходимо учитывать при их синтезе. Ионы MnO4n– представляют собой правильные тетраэдры со значительной долей π-связывания, заключающегося в перекрывании заполненных р-орбиталей атомов кислорода со сходными по симметрии d-орбиталями марганца. Степень π-связывания , а следовательно, и кратность сязи Mn–O возрастает по мере увеличения степени окисления марганца, что приводит к уменьшению длины связи Mn–O от 0.171 нм в MnO43– до 0.166 нм в MnO42– и 0.163 нм в MnO4–. Все оксоманагнаты имеют яркую окраску, зависящую от степени окисления: гипоманганаты – синие, манганаты – зеленые, перманганаты – фиолетовые. Хотя в ионах MnO43– и MnO42– содержатся неспаренные электроны, окраска всех трех типов оксоманаганганатов обусловлена не d-d переходами, а интенсивной полосой переноса заряда. Перманганаты щелочных металлов изоморфны перхлоратам, манганаты – сульфатам и хроматам, а гипоманганаты – ортофосфатам и ванадатам. Эти соли можно использовать в качестве матриц для стабилизации неустойчивых ионов.
Гипоманганаты присутствуют в продуктах высокотемпературарного термического разложения перманганатов или манганатов:
500-850 оС:
3K2MnO4 = 2K3MnO4 + MnO2 + O2↑,
а также окисления пиролюзита кислородом в сильно щелочной среде:
300 оС
2MnO2 + 6КОН + 1/2О2 = К3MnO4 + 3Н2О.
Полученные таким образом, они представляют собой сине-зеленые кристаллические порошки, устойчивые только в отсутствии следов влаги из-за легкости диспропорционирования:
2MnO43- + 2Н2О = MnO42- + MnO2 + 4ОН-
На сухом воздухе они выдерживают нагревание до 900 оС. В растворе их удается стабилизировать только на холоду и в сильно щелочной среде (константа скорости реакции диспропорционирования составляет 6.9 л/моль–1×с–1 в 3М KOH и > 104 л/моль–1×с–1 при рН = 12 и 20°C). Например, К3MnO4 при –15 оС растворяется в холодном 40 %-ом гидроксиде калия, образуя ярко-синий раствор, из которого могут быть получены кристаллогидраты голубого цвета. Однако уже при 0 оС эти растворы подвергаются диспропорционированию. Небесно-голубые кристаллы соли Na3MnO4×0,25NaOH×12H2O, синтез которой приведен в практических руководствах:
2KMnO4 + 2Na2SO3 + 6NaOH + 2H2O = 2Na3MnO4 + 2Na2SO4 + 2KOH
несмотря на частичную стабилизацию гидроксид-ионами, разлагаются в течение нескольких часов.
Неустойчивые голубые растворы, по-видимому, содержащие гипоманганат-ионы в протонированной форме HMnO42–, удается получить при восстановлении манганата калия в сильно-щелочной среде (10 M KOH) пероксидом водорода или формиатом натрия (J.D. Rush, B.H.J. Bielski, Inorg. Chem., 1995, 34, 5832; D.G. Lee, T. Chen, Journ. Amer. Chem. Soc., 1989, 111, 7535). При этом к охлажденному щелочному раствору манганата восстановитель надо добавлять по каплям. Удобным способом увидеть красивую ярко-голубую окраску гипоманганата является восстановление перманганата в расплавленной щелочи. Этот способ, разработанный одним из авторов книги, до сих пор не описан в литературе, поэтому мы приводим методику эксперимента. В сухую пробирку положите десяток гранул твердого гидроксида калия и добавьте к ним одну каплю раствора перманганата калия. Нагрейте пробирку на пламени. Сначала расплав стает изумрудно-зеленым вследствие образования манганата, а при дальнейшем нагревании – ярко-голубым. При охлаждении он затвердеет, сохраняя окраску. Гипоманганат-ионы оказывают стабилизированными в структуре твердой щелочи.
Разложение безводных гипоманганатов протекает лишь при высоких температурах, и его направление зависит от природы катиона. Так, натриевая соль выше 1250 оС вследствие выделения летучего оксида натрия отщепляет кислород, превращаяясь в MnO
Калийная соль при разложении дает оксоманганаты(III) или смешанную фазу K2Mn4O7,8, в которой марганец находится в двух степенях окисления. На влажном воздухе при 250 оС K3MnO4 окисляется:
2K3MnO4 + 1/2О2 + H2O= 2K2MnO4 +2КОН.
Оксоманганат (V) Li3MnO4, полученный в токе кислорода по реакции:
120 оС
LiMnO4 + 2LiOH = Li3MnO4 + H2O + 1/2O2↑
при мольном соотношении перманганата калия и гидроксида лития, взятого в форме моногидрата, 1 : 3.1, термически менее устойчив, чем калийная и натриевая соли.
Впервые манганаты(VI)приготовил Глаубер в 1659 г. при растворении пиролюзита в расплавленной селитре. Эти вещества представляют собой темно-зеленые, почти черные кристаллы, растворимые в растворах щелочей с образованием изумрудно-зеленых растворов. Магнитный момент составляет 1,78-1,84 М.Б., что соответствует одному неспаренному электрону d1. В нейтральных и слабокислых растворах манганаты легко диспропорционируют:
3K2MnO4 + Н2О = 2KMnO4 + MnO2↓ +4КОН,
о чем судят по выпадению черного осадка диоксида и появлению малинового окрашивания. Для ускорения процесса обычно прибегают к пропусканию через раствор углекислого газа:
3K2MnO4 + 2СО2 = 2KMnO4 + MnO2↓ + 2К2СО3
или добавляют соль аммония:
3K2MnO4 + 4NH4Cl = 2KMnO4 + MnO2↓+ 4NH3 + 4KCl + 2H2O.
Среди манганатов наибольшее значение имеет калийная соль K2MnO4, служащая для производства перманганата KMnO4. Ее синтезируют окислением пиролюзита в присутствии кислорода в расплаве щелочи или в ее концентрированном растворе
MnO2 + 2KOH + 1/2O2 = K2MnO4 + H2O.
В качестве окислителей используют также хлорат калия и нитрат калия или их смесь:
3MnO2 + 6KOH + KClO3 = 3K2MnO4 + KCl + 3H2O,
MnO2 + 2KOH + KNO3 = K2MnO4 + KNO2 + H2O.
Окисление этими реагентами с заметной скоростью начинается при 170 оС. Продукт извлекают водой, и прозрачный раствор кристаллизуют. При этом часть вещества теряется из-за диспропорционирования.
Этот процесс протекает в несколько стадий. Сначала реакция не сопровождается изменениями степени окисления марганца:
MnO2 + 2КОН = K2MnO3 + Н2О.
Затем, при расплавлении щелочи (380 оС), наступает быстрое окисление MnО2:
2MnO2 + 6КОН + 1/2О2 = K3MnO4 + Н2О.
Для проведения последующей стадии требуется понижение температуры ввиду более низкой термической устойчивости манганата по сравнению с гипоманганатом:
K3MnO4 + Н2О + 1/2О2 = K2MnO4 + 2КОН.
Максимальный выход K2MnO4 достигается при 190-210 оС. Помимо манганита и гипоманганата, в качестве промежуточных продуктов зафиксированы также K2Mn8O16, K2Mn4O8 и K2Mn2O(4-δ). При синтезе манганата из пиролюзита в жидкой фазе процесс проводят в 40 %-ом растворе КОН при температуре 150-160 оС и давлении кислорода 100 атм. Продукт кристаллизуют и промывают 60 %-ным раствором гидроксида калия при 100 оС.
В электрохимическом методе синтеза пиролюзит используют в качестве анода. Катодом служит железная трубка, электролиз проводят в 90 %-ном КОН при 200 оС. На ряде производств анодному окислению подвергают металлический марганец без примеси железа, при этом образуется смесь перманганата и манганата с выходом 80 %. Повышая температуру процесса и концентрацию щелочи, удается увеличить количество манганата.
В качестве окислителей при синтезе манганатов могут быть использованы пероксиды и азиды:
BaO2 + MnO2 = BaMnO4,
4MnO2 + KN3 + 7KOH + H2O = 4K2MnO4 + 3NH3↑.
В лабораторных условиях манганаты щелочных металлов удобно получать окислением воды перманганатом в сильно щелочном раcтворе. Для этого перманганат помещают в концентрированный раствор щелочи и нагревают:
2NaMnO4 + 2NaОН = 2Na2MnO4 + Н2О + 1/2О2↑.
В качестве восстановителя может быть использован амид калия в жидком аммиаке:
6KMnO4 + 6KNH2 = 6K2MnO4 + 4NH3↑ + N2↑.
Манганаты термически менее устойчивы, чем гипоманганаты: K2MnO4 разлагается при 600 оС, выделяя кислород.
Для синтеза манганатов рубия, цезия и бария используют обменные реакции:
Na2MnO4 + Ba(OH)2 = BaMnO4↓ + 2NaOH.
При увеличении степени окисления марганца термическая устойчивость оксоманганатов уменьшается, а их окислительное действие усиливается, что, например, следует из значений стандартных электродных потенциалов. Практического значения манганаты и гипоманганаты не имеют, так как в водных растворах легко диспропорционируют, давая перманганат-ионы.
Малиново-фиолетовые растворы, содержащие перманганат-ионы MnO4–, также образуются при окислении солей марганца(II) оксидом свинца(IV), висмутатом натрия, персульфатом аммония, иодной кислотой и другими сильными окислителями:
2Mn(NO3)2 + 5PbO2 + 6HNO3 = 2HMnO4 + 5Pb(NO3)2 + 2 H2O,
2Mn(NO3)2 + 5NaBiO3 + 16HNO3 = 2HMnO4 + 5Bi(NO3)3 + 5NaNO3 + 7H2O,
2MnSO4 + 5(NH4)2S2O8 + 8H2O = 2HMnO4 + 5(NH4)2SO4 + 7H2SO4.
Эти реакции проводят в кислой среде, так как в этих условиях перечисленные выше вещества в наибольшей степени проявляют окислительные свойства.
Для проведения реакций берут очень немного соли марганца, так как в избытке происходит ее сопропорционирование с образующимся перманганатом. Таким образом, эти превращения не могут применяться для синтеза перманганатов, а служат лишь удобным аналитическим методом определения ионов марганца(II). При использовании в качестве восстановителя персульфата требуется присутствие катализатора – ионов Ag+. Механизм реакции полностью не изучен, однако известно, что лимитирующей стадией этого процесса является окисления персульфатом ионов серебра, возможно, в форме комплекса
S2O82– + 2Ag+ = 2Ag2+ + 2SO42–
которые и осуществляют последовательное окисление марганца(II) до перманганата. Аналогично персульфату реагирует пероксофосфат P2O84– (Jagan Mohana Rao K., Vittal A.S.P., Krishna Rao P.V. Inorg. Chem., 1984, 23, 3212)
Перманганаты лития, натрия, кальция и стронция сильно гигроскопичны и образуют ряд кристаллогидратов, например, LiMnO4.3H2O, NaMnO4.3H2O, NaMnO4.H2O (Рис.5.35. Фазовая диаграмма системы NaMnO4 – H2O). Растворимость перманганатаов щелочных металлов убывает с ростом катиона, соль лития является исключением (Табл.5.4).
Таблица 5.4. Растворимость некоторых перманганатов (г безводной соли в 100 г воды) при 20 °C
| Катион | Li | Na | K | Rb | Cs | Ca | Sr | Ba | NH4 | Ag |
| Растворимость соли | 32,5 | 59,0 | 6,3 | 1,1 | 0,23 | 62,5* | 7,4** | 0,92 |
* - при 11°C, ** - при 15°C.
Перманганат аммония начинает разлагаться уже при 70°C, а при ударе или нагревании до 110 оС взрывается (L. Kotai, P. Szabo, A. Keszler, Thermochim. Acta, 1999, 338, 129):
2NH4MnO4 = 2MnO2 + H2O + 3/2O2↑ + 2NH3↑.
Перманганат рубидия при нагревании ведет себя так же, как и калийная соль. Разложение гидрата перманганата кальция приводит к отщеплению кристаллизационной воды и последующему разложению соли при 200 °C с общей потерей массы, соответствующей образованию шпинели CaMn2O4. Бариевая соль разлагается при 220 °C.
Синтез перманганатов в лаборатории проводят окислением манганатов хлором, бромом или периодатом в щелочной среде:
2K2MnO4 + Cl2 = 2KMnO4 + 2KCl.
Реакция протекает количественно. Из сильно концентированных растворов сразу выпадают кристаллы. Окисление манганатов в щелочных растворах ионами ClO– или H3IO62– включает диспропорционирование:
2MnO42– = MnO43– + MnO4–
и окисление возникающего гипоманганата:
MnO43– + H3IO62– = MnO4– + IO3– + 3OH-.
Окисление озоном не находит практического применения:
2K2MnO4 + О3 + Н2О = 2KMnO4 + 2КОН + О2↑.
Другими способами являются диспропорционирование манганатов, а также обменные реакции между солями:
KMnO4 + NH4Cl = NH4MnO4↓ + KCl (осаждается при охлаждении),
AgMnO4 + NaCl = NaMnO4 + AgCl↓.
Таким же способом удается получить и растворы марганцевой кислoты НМnO4:
Ba(MnO4)2 + H2SO4 = BaSO4↓ + 2HMnO4,
2KMnO4 + H2SiF6 = K2SiF6↓ + 2HMnO4.
Марганцевая кислота относится к сильным кислотам, поэтому перманганаты щелочных металлов практически не гидролизуются. Разбавленные свежеприготовленные водные растворы марганцевой кислоты можно нагревать до кипения без заметного разложения. Однако, при длительном хранении водные растворы постепенно разлагаются с образованием осадка MnO2 и выделением кислорода:
4НМnO4 = 4MnO2↓+ 3О2↑ + 2Н2О.
Марганцевая кислота известна только в водных растворах.
Выпариванием получен насыщенный при 18 оС раствор с концентрацией 1,4 моль/литр (17 г/100 мл раствора). Растворением марганцевого ангидрида в воде достигнуты концентрации кислоты до 20 %. Полное разложение 0,3 М раствора при комнатной температуре происходит за два месяца. При попытках выделения твердой кислоты вымораживанием растворов получены кристаллы (H3O)2[Mn(MnO4)6].11H2O, в состав которых входят атомы марганца в разных степенях окисления, Mn(IV) и Mn(VII) (B.Krebs, K.D.Hasse, Angew. Chem., 1974, 86, 647). Имеются сведения о выделении кристаллов безводной кислоты HMnO4 и ее оксониевой соли H5O2+MnO4–.
Соли марганцевой кислоты – одни из важнейших неорганических окислителей, активные в твердой фазе и лабильные в растворе. С их помощью можно окислить многие органические и неорганические вещества. Такой широкий спектр действия объясняется сложным механизмом окиления, включающим несколько параллельных путей, хотя бы один из которых практически всегда оказывается возможным, а высокая скорость протекания процессов обусловлена низкой энергией активации – от 21 до 42 кДж/моль. Наиболее сильными окислительными свойствами перманганаты обладают в кислотной среде, где они восстанaвливаются до ионов Mn2+:
MnO4– + 8H+ + 5e– ® Mn2+ + 4H2O, E0 = 1,51 B (pH = 0).
В нейтральной и слабощелочной среде перманганат восстанавливается до диоксида марганца, выпадающего из раствора в виде бурого осадка
MnO4– + 2H2O + 3e– ® MnO2 + 4OH–, E0 = 1,15 B (pH = 7).
а в сильнощелочной (рН > 12,5) образуется изумрудно-зеленый манганат, устойчивый при рН > 14:
MnO4– + e– ® MnO42– , E0 = 0,56 B (pH = 14).
Хотя механизмы многих реакций с участием перманганат-иона являлись предметом тщательного изучения, однозначного вывода о протекании процессов сделать не удается. Механизм восстановления перманганат-иона включает несколько стадий, представляющих собой одно- и двухэлектронные переносы. В качестве интермедиата в щелочной среде практически всегда удается зафиксировать гипоманганат, дающий сигналы в спектре электронного парамагнитного резонанса и характерный электронный спектр (Рис.5.36. Электронные спектры перманагната калия и продуктов его восстановления, включая промежуточные соединения; 20 °C, 0.2 ммоль/литр). Приведем предполагаемый механизм реакции взаимодействия перманганата с сульфитом при рН = 9 (L.I. Simandi, M. Jaki, C.R. Savage, Z.A. Schelly, Journ. Amer. Chem. Soc., 1985, 107, 4220; 1984, 106, 6866). Первой стадией процесса являются два параллельных внешнесферных одноэлектронных переноса, приводящих к окиcлению сульфит-иона до сульфата:
MnO4– + SO32– ® MnO42– + SO3–
MnO4– + SO3– + 2OH– ® MnO42– + SO42– + H2O.
Образующийся манганат-ион является более слабым и более медленно действующим окислителем, чем перманганат, поэтому он успевает претерпеть диспропорционирование, которое приводит к гипоманганату:
2MnO42–® MnO4– + MnO43–
Гипоманганат, в свою очередь, также диспропорционирует на перманганат и диоксид марганца, который сначала образует коллоидный раствор желтого цвета, а лишь спустя некоторое время осаждается:
2MnO43–® MnO2(sol) + MnO4–,
MnO2(sol) ®MnO2¯.
Суммарное уравнение имеет вид:
2MnO4– + 3SO32– + H2O ® 2MnO2¯ + 3SO42– + 2OH–.
Восстановление перманганатом сульфита в кислой среде протекает через интермедиаты - соединения марганца(III). Их удалось стабилизировать в виде K2[MnF3SO4]и K2[MnF5], введя в раствор фторид-ионы (C. Bhattacharyya et al, Journ. Chem. Soc., Dalton Trans., 1993, 4397).
С органическими веществами, например, спиртами или олефинами, перманганат-ион взаимодействует по внутрисферному механизму через хелатный цикл, внутри которого и происходит двухэлектронный перенос. Дальнейший механизм определяется рН раствора. В сильнощелочной среде манганат(V) достаточно устойчив к диспропорционированию, поэтому окисление происходит под действием другого перманганат-иона. В среднещелочных, нейтральных и кислотных растворах протекает диспропорционирование. Конечный продукт реакции, диол, образуется в результате гидролиза хелатного комплекса, а манганат-ионы, диспропорционируя, переходят в диоксид марганца ().D. G. Lee, T. Chen, Journ. Amer. Chem. Soc., 1989, 111, 7534).
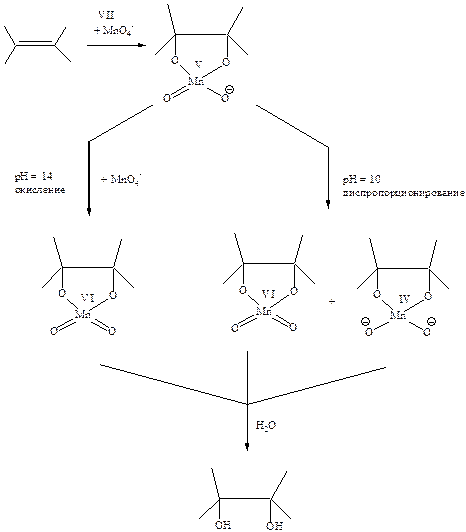
К недостаткам перманганатов как окислителей следует отнести невозможность их использования в органических растворителях, по причине их низкой растворимости в малополярной среде. Однако это удается преодолеть при использовании межфазного катализа, например, добавляя краун-эфиры (см. т.2, стр 46).
ДОПОЛНЕНИЕ. Перманганат калия.
Перманганат калия представляет собой фиолетово-черные блестящие гигроскопичные кристаллы, растворимые в воде с образованием ярко окрашенных малиновых растворов. Насыщенный раствор этой соли (растворимость в 100 г воды: 6,4 г при 20°C, 22,2 г при 60°C, Рис.5.37. Кривая растворимости перманганата калия в воде; координаты эвтектики: 2,91%, –0,58°C) почти черного цвета. В кристаллическом перманганате KMnO4 атомы марганца располагаются в правильных тетраэдрах MnO4, в которых межатомные расстояния составляют: Mn-O 0,162 – 0,163 нм, а углы О-Mn-O 108,9-111,0 о. Связи Mn-O ковалентные. Перманганат калия изоморфен с перманганатами рубидия, цезия, аммония, а также с перхлоратами калия и таллия(I), сульфатами бария и стронция, хроматом бария, метапериодатом и тетрафтороборатом калия. Интересно, что он не образует смешанных кристаллов с перманганатом натрия. Недавно получены смешанные кристаллы манганата-перманганата калия K3Mn2O8 (M.B. Hursthouse et al, Journ. Chem. Soc., Faraday Trans., 1992, 88, 3071 ).
При нагревании перманганат разлагается с выделением кислорода:
220 °C
2KMnO4 = K2MnO4 + MnO2 + О2↑.
Дальнейший ход разложения представлен на рис.5.38 (Рис.5.38 Кривая потери массы при разложении перманганата калия на воздухе, скорость нагрева 10 °/мин, материал тигля - алунд. Метод изучения изменения массы вещества при нагревании, называют термогравиметрическим анализом, а представленную кривую – дериватограммой).
Выше 100 оС он восстанавливается водородом:
10KMnO4 + 10Н2 = 5K2MnO4 + 5MnO2 + 10H2O.
Смеси KМnO4 с серой, фосфором, мышьяком, сурьмой, порошками кремния, алюминия, титана при растирании или нагревании загораются, при соприкосновении кристаллов перманганата с жидкой серой происходит взрыв. Смесь тонкого порошка углерода с перманганатом может служить для зажигания. Все это свидетельствует о высокой окислительной активности перманганата. В водных растворах он легко окисляет сульфиты, сульфиды, иодиды, оксалаты и многие другие типичные восстановители, а также органические вещества, содержащие связи С=С, СºС, С–ОН, С=О.
При растворении перманганата калия в олеуме образуется зеленый раствор
KMnO4 + 3H2S2O7 = K+ + MnO3+ + HSO4– + HS3O10– + 2H2SO4,
из которого через 2-3 часа выделяется кислород и выпадает MnO2. Раствор KMnO4 в азотной кислоте имеет интенсивный красный цвет, и из него также постепенно выделяется осадок диоксида. Этим пользуются для получения чистого пиролюзита. При обработке перманганата фосфорной кислотой (95-100 %) выделяется Mn2O7. С безводной HF и фторосерной кислотой HSO3F перманганат дает зеленый раствор и пары зеленого цвета, из которых выделен зеленый MnO3F. Интересно, что перманганат калия не растворим в жидком хлороводороде, хотя газообразный HCl или его водный раствор легко окисляются им до свободного хлора.
При 40 оС пентафторид иода взаимодействует с KMnO4 как фторирующий агент:
KMnO4 + IF5 = MnO3F + IOF3 + KF.
Перманганат катализирует термическое разложение хлората калия и перхлората аммония. Благодаря сильным окислительным свойствам, он используется как окислитель при подготовке и очистке воды и воздуха, служит эффективным антисептиком в фармакологии, как реагент в аналитической и органической химии.
Перманганат калия синтезируют электролитическим окислением манганата K2MnO4 в щелочных растворах или анодным окислением марганца и его сплавов:
2K2MnO4 + 2H2O = 2 KMnO4 + 2KOH + H2↑.
Для устранения катодного восстановления MnO2 процесс проводят с диафрагмой. Исходный раствор содержит 150-250 г/литр K2MnO4 и 100-250 г/литр КОН. За время 10-40 часов при 30-65 оС окисляется 70-95 % K2MnO4. Аналогично протекает анодное окисление манганата натрия Na2MnO4 до перманганата.
Анодное окисление ферромарганца протекает по уравнению:
Mn + OН- + 3Н2О = MnO4- + 3,5 Н2↑.
В процессе электролиза перманганат-ионы образуются на аноде.
При использовании разбавленных растворов щелочи или поташа происходит образование перманганата калия, тогда как при высоких концентрациях щелочи выделяется манганат K2MnO4. При охлаждении раствора с рН 7,5 –10, насыщенного при 60 оС, выпадают кристаллы KMnO4 размером до 4 мм. Всегда присутствующий в водных щелочных растворах перманганата диоксид марганца коагулируют введением до 1 % поверхностно активного вещества и тем самым стабилизируют эти растворы. Аналитически чистый продукт получают двойной перекристаллизацией.
Дата добавления: 2016-01-03; просмотров: 3249;