Лечение 1 страница
и-
\ Лечение хирургическое, направленное на увеличение объёма погости черепа.
)■ Принципы хирургической реконструкции верхней зоны лица и ча-^ти свода черепа одинаковы для хирургического лечения всех форм ЭКраниосиностозов. Методы заключаются в выдвижении или реконструкции верхнеглазничного блока и создании «плавающего» лба. рГаким образом создают условия для формирования лицевого и мозгового черепа в послеоперационном периоде в правильных анатомических пропорциях. Для реконструкции верхней зоны лица исполь-1уют следующие методы:
| 1) выдвижение фронто-супраорбитального фрагмента единым Шоком;
к 2) реконструкция супраорбитального блока с последующим его выдвижением;
3) создание костного мостика;
4) создание крыловидного костного мостика.
Реконструированную тем или иным способом лобную кость фик-
сируют к реконструированным структурам верхней зоны лица.
Фиксация лобной кости на выдвинутом фрагменте верхних лицевых структур позволяет создать значительные костные дефекты между краями лобной кости и остальными костями свода черепа, что даёт возможность избежать рецидива краниосиностоза и краниостеноза.
Эти методы применяют при выполнении первого этапа хирургического лечения краниофациальных дизостозов Крузона, Апера, Пфайффера и Сетре—Хотцена.
Коррекция тригоноцефалии
Тригоноцефалия, или метопический синостоз, занимает первое место по количеству наблюдаемых больных с синостозами. Для этой формы синостоза характерна килевидная деформация лба с наличием костного гребня по ходу синостозированного метопического шва (рис. 3-13).
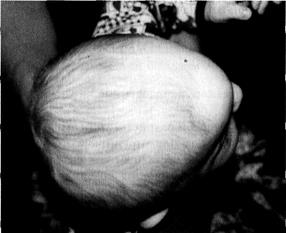
Рис. 3-13. Внешний вид ребёнка с тригоноцефалией.
За счёт расширения черепа в поперечной плоскости по теменно-височному размеру и его сужения в лобной области череп приобретает грушевидную форму. У большинства больных с тригоноцефалией выявляют гипотелоризм — близкое стояние орбит. Тригоноцефалию чаще встречают у мальчиков.
Объём хирургического вмешательства зависит от степени выраженности тригоноцефалии и возраста ребёнка. У детей в возрасте от 6 до 9 мес порок развития заключается только в значительном уплотнении метопического шва, он контурируется под кожей, при этом нет западения лобно-височных областей. В этих случаях можно ограничиться реконструкцией лобной кости.
Коррекция плагиоцефалии
Этот вид синостоза занимает второе место по частоте после тригоноцефалии. Он характеризуется преждевременным закрытием одной половины венечного шва и (в большинстве случаев) за-ращением швов передней черепной ямки (сфено-фронтального и фронто-этмоидального). В результате этого возникают фронто-темпоральная и скуловая деформации. У детей в более позднем возрасте (6—9 лет) развивается деформация лица: искривление пирамиды носа и носовой перегородки, асимметрия ушных раковин (рис. 3-14).
Применяют три модификации исправления лобной плагиоцефалии в зависимости от степени выраженности этого синостоза.

При отсутствии компенсаторного выбухания лобно-теменной области выполняют костно-пластическую резекцию лобной кости на стороне поражения. При наличии такой деформации проводят тотальную резекцию лобной кости. Независимо от степени выраженности деформации все операции выполняют в три этапа. • I этап. Распилы в поражённой зоне лобной кости выполняют по линии отсутствующего венечного шва параллельно надбровной дуге
и лобно-височной области. Фрагмент лобной кости снимают и помещают в стерильную ёмкость с физиологическим раствором.
• II этап. Выпиливание супраорбитального блока выполняют по линиям лобно-носового и лобно-скулового швов, на основании черепа — по линии отсутствующих сфено-фронтального и сфено-этмо-идального швов.
• III этап операции заключается в реконструкции лобной кости на стороне поражения. Цели этого этапа — достижение наиболее близкой к физиологической кривизны лобной кости и формирование лобного бугра.
Техника операции при затылочной (задней) плагиоцефалии
Наиболее удобное положение больного — на животе. Хирургический подход отличается тем, что кожный разрез делают ближе к затылочной области.
В области западения затылочной кости резецируют костный фрагмент округлой формы. Реконструкцию проводят путём радиальной остеотомии по типу «лепестков ромашки». Получившиеся костные язычки надламывают у основания. Таким образом достигают увеличения округлости контуров затылочной области.
Вторая методика заключается в выкраивании лоскута с помощью выкусывания костной «дорожки» по верхнему и боковым контурам зоны западения. Затем этот лоскут надламывают у основания и отводят кнаружи.
Коррекция скафоцефалии
Скафоцефалия (сагиттальный синостоз) в большинстве своём поражает мальчиков (до 85% случаев). Эта форма краниосиностоза занимает третье место. Для этой формы синостоза характерны сужение черепа в поперечной плоскости и компенсаторное его вытяжение в передне-заднем направлении. Череп принимает ладьевидную форму (рис. 3-15).
Существует несколько методик коррекции этого краниосиностоза. В частности, выпиливание поочерёдно теменных, лобной и затылочной костей. Затылочную кость резецируют до уровня поперечного венозного синуса. Над сагиттальным синусом в результате резекции костей свода черепа остаётся небольшая полоска. Реконструкцию костей выполняют с помощью клиновидных радиальных распилов по типу «лепестков ромашки». Это даёт возможность в значительной
степени уменьшить выпуклость лобной и затылочной костей. Для уменьшения передне-заднего размера черепа выполняют резекцию костных фрагментов треугольной формы в супраорбитальной области с обеих сторон.
Коррекция туррицефалии
Это деформация черепа развивается при заращении венечных швов с обеих сторон. Череп приобретает вытянутую по вертикали форму (башенный череп). Происходит ещё и срастание швов основания черепа. Профиль черепа у таких детей имеет отличительную особенность: линия носа и линия лба составляют единую прямую линию (рис. 3-16).
При рентгенографическом исследовании определяют башнеобразную деформацию, истончение костей черепа, множественные пальцевые вдавления, характерные для синдрома внутричерепной гипер-тензии, укорочение размеров основания черепа (рис. 3-17).
Хирургическую коррекцию туррицефалии выполняют по следующей методике: на первом этапе проводят резекцию лобной кости, верхнеглазничный блок резецируют, его височный фрагмент выполняют в виде конуса, чтобы изменить угол наклона блока. Затем формируют костный мостик.
Лобную кость распиливают на горизонтальные фрагменты. Их кривизну изменяют с помощью щипцов Мюллера. Затем фрагменты

лобной кости устанавливают в порядке, обеспечивающем наиболее эстетически приемлемую кривизну лобной кости.
Такая методика позволяет в значительной степени изменить очертания профиля, уменьшить вертикальные размеры свода черепа и удлинить его основание.
Хирургическое лечение краниостеноза
Крайнее проявление краниосиностозов — краниостеноз. Эта стадия характеризуется истощением компенсаторных возможностей ЦНС в условиях повышенного внутричерепного давления. Основная задача хирурга в этой ситуации — устранение сдавления головного мозга костями свода черепа. Наиболее распространённая операция — двухлоскутная краниотомия.
Суть вмешательства заключается в выкусывании параллельных дорожек в теменно-височных областях свода черепа и резекции надкостницы на протяжении 2 см от края области остеоэктомии.
Для большего увеличения объёма полости черепа выполняют че-тырёхлоскутную краниотомию.
Коррекция орбитального гипертелоризма
Часть краниосиностозов сочетается с орбитальным гипертелоризмом — врождённым состоянием, вторичным по отношению к основному врождённому пороку развития. Термин «гипертелоризм» происходит от греческих слов hyper (сверх), tele (далеко) и horismos (разграничение). Гипертелоризм — ненормально большое расстояние между глазницами (рис. 3-18).
Существуют два основных метода хирургической коррекции этого состояния: подчерепная остеотомия и коррекция орбитального гипертелоризма внутричерепным доступом. Эти методы позволяют за счёт перемещения резецированных костных пластинок черепа исправить Деформацию и приблизить анатомию лицевого скелета к норме.
Подводя итог сказанному, следует отметить, что неукоснительное соблюдение принципов хирургического лечения краниосиностозов и орбитального гипертелоризма позволяет добиться максимального косметического и функционального эффектов. А это, в свою очередь, служит основой для полной медицинской и социальной реабилитации больных детей.
3.2.4. Спинномозговая грыжа
Спинномозговая грыжа — тяжёлый порок развития, характеризующийся врождённым незаращением задней стенки позвоночного канала с одновременным грыжевым выпячиванием твёрдой мозговой оболочки, покрытым кожей и содержащим спинномозговую жидкость либо спинной мозг и/или его корешки. Комплекс анатомических и функциональных нарушений, возникающий при спинномозговой грыже, условно объединяют термином «миело-дисплазия».
Миелодисплазия — собирательное понятие, объединяющее большую группу пороков развития спинного мозга с типичной локализацией (синдром каудальной регрессии) и широким диапазоном проявлений, от рахишизиса до скрытых, сугубо тканевых изменений. Миелодисплазию подразделяют на две формы: органную (спинномозговые грыжи) и тканевую (к костным маркёрам последней относят незаращение дужек позвонков, агенезию крестца и копчика, диастематомиелию и др.). Косвенные признаки тканевой формы миелодисплазии: участки кожной пигментации или депигментации в пояснично-крестцовой области, очаги интенсивного оволосения, дермальный синус в верхней части межъягодичной складки.
Частота рождения детей со спинномозговой грыжей составляет 1 случай на 1000—3000 новорождённых. Порок развития на уровне шейного отдела позвоночника встречают в 3,2% случаев, грудного — в 18%, люмбосакрального — в 56%. Около 40% составляет корешковая форма {meningoradiculocele).
Достаточно часто эта патология сочетается с другими врождёнными аномалиями: гидроцефалией, диастематомиелией, синдромами Арнольда—Кияри и Клиппеля-Фейля, краниостенозом, незаращением верхней губы, кардиопатией, пороками развития нижних мочевых путей, атрезией ануса.
При врождённых мальформациях люмбосакрального отдела позвоночника (спинномозговой грыже, агенезии крестца и копчика, различных вариантах дисплазии люмбосакрального сочленения, диастематомиелии и др.) на фоне разнообразных неврологических проявлений (недержания мочи и кала, снижения тонуса мышц тазового дна, нижних вялых параличей и парезов, трофических язв промежности и нижних конечностей) часто встречают варианты регионарного тканевого дисморфизма в виде геманги-ом, липом, липофибром и лимфангиом («синдром спинального диз-рафизма»).
В основе возникновения спинномозговой грыжи лежит порок развития спинного мозга, появляющийся как результат задержки закладки и замыкания медуллярной пластинки в мозговую трубку. Нарушается процесс развития и замыкания дужек позвонков, который в норме совместно с образовавшимися из эктодермы мягкими тканями и оболочками мозга, получившими начало из мезодермы, закрывает спинномозговой канал. В результате наличия костного дефекта задней стенки позвоночного канала под влиянием повышенного давления ликвора в субарахноидальном пространстве спинной мозг с корешками и оболочками выпячивается, формируя объёмное образование, частично или полностью покрытое кожей.
Этиология этого порока развития изучена недостаточно. Большое количество физических, химических и биологических факторов, действуя на организм плода в период формирования позвоночника, могут стать причиной этой врождённой мальформации. Варианты кау-дального дизэмбриогенеза представлены на рис. 3-19.
Классификация
В зависимости от степени недоразвития и участия отдельных элементов спинного мозга и позвоночника в патологическом процессе различают несколько анатомических форм (рис. 3-20).
Менингоцеле
При этой форме порока выявляют незаращение дужек позвонков. Через дефект выпячиваются только оболочки спинного мозга. Содержимое грыжевого мешка — спинномозговая жидкость без элементов нервной ткани. Спинной мозг обычно не изменён и расположен

правильно. В неврологическом статусе возможны разнообразные варианты нарушений функции тазовых органов, явления дистально-го парапареза, паретические деформации стоп.
Менингорадикулоцеле
В состав грыжи входят корешки спинного мозга, часть которых сращена с внутренней стенкой грыжевого мешка. При этой форме порока развития часто наблюдают дополнительные интрамедулляр-ные образования в виде липом (липоменингоцеле), врождённых арах-ноидальных кист и т.д. Спинной мозг имеет обычное анатомическое расположение. Степень выраженности неврологической симптоматики зависит от уровня поражения. Чем выше локализация дефекта позвоночника, тем более выражены неврологические «выпадения» со стороны органов малого таза и нижних конечностей.
Менингомиелоцеле
При этой форме помимо оболочек и корешков спинного мозга в грыжевое содержимое вовлечён непосредственно и спинной мозг. Обычно спинной мозг, выйдя из спинномозгового канала, проходит в грыжевой мешок и заканчивается в его центре в виде не замкнувшейся в трубку зародышевой мозговой пластинки. Серое и белое вещество этого участка спинного мозга сформировано неправильно. При этой форме порока развития значительно выражены неврологические дефекты: у детей обычно выявляют тотальное недержание мочи и кала, вялый или спастический парапарез, паретическую деформацию нижних конечностей.
Миелоцистоцеле
Миелоцистоцеле — самая тяжёлая форма грыж. При ней спинной мозг страдает особенно сильно, выпячиваясь вместе с оболочками через дефект позвоночника. Истончённый спинной мозг растянут спинномозговой жидкостью, скапливающейся в порочно расширенном Центральном канале, нередко мозг прилегает к внутренней стенке грыжевого мешка или сращён с ней. Для этой формы характерны тяжёлые неврологические расстройства с нарушениями функций тазовых органов и парезом нижних конечностей. Миелоцистоцеле может располагаться в шейном, грудном и грудопоясничном отделах
позвоночника. Наиболее тяжёлые и необратимые неврологические выпадения регистрируют именно при этой форме порока развития.
Рахишизис
При этой патологии происходит полное расщепление мягких тканей, позвоночника, оболочек и спинного мозга. Спинной мозг, не сомкнувшийся в трубку, лежит в виде бархатистой массы красного цвета, состоит из расширенных сосудов и элементов мозговой ткани. Задний рахишизис нередко сочетается с передним (когда расщеплены не только дужки, но и тела позвонков) и тяжёлыми уродствами головного мозга и других органов. Наиболее часто рахишизис встречают в поясничной области. Дети с этой формой порока развития нежизнеспособны.
Spina bifida occulta
Spina bifida occulta — скрытый дефект дужек позвонков, когда отсутствует грыжевое выпячивание. Наиболее частая локализация этой формы — крестцовый и поясничный отделы позвоночника. На уровне незаращения дужек позвонков можно наблюдать различные патологические образования в виде плотных фиброзных тяжей, хрящевой и жировой ткани, липом, фибром и др. При рентгенографии выявляют незаращение дужек, иногда и тел позвонков. Незаращение дужек позвонков, осложнённое опухолью (например, липомой, фибромой), известно под названием spina bifida complicata. Липоматозная ткань располагается под кожей, заполняет дефект в дужках позвонков и может не только срастаться с оболочками спинного мозга, но и проникать в субарахноидальное пространство, где нередко интимно срастается с корешками и спинным мозгом, расположенным ниже обычного уровня (интрарадикулярный рост).
Диастематомиелия
Диастематомиелия — аномалия позвоночника, характеризующаяся наличием костного шипа (у части больных инверсия остистого отростка), сдавливающего и разделяющего спинной мозг на две половины. Обычно аномалия имеет множество дополнительных дизра-фических признаков в виде гиперпигментации кожи и гипертрихоза над зоной поражения, короткой терминальной нити, сколиоза и т.д.
Клиническая картина
По средней линии позвоночника, обычно в поясничном отделе, определяют опухоль (размеры варьируют), покрытую часто истончённой в центре и рубцово-изменённой кожей. При резком истончении кожи опухоль просвечивает. У её основания бывает расположен участок чрезмерного оволосения или сосудистое пятно. У основания опухоли пальпируют несросшиеся дужки позвонков (рис. 3-21).
Расстройства чувствительности при врождённых пороках спинного мозга складываются из сегментарных, проводниковых и корешковых нарушений. Они могут проявляться анестезией, гипестезией, реже — гиперестезией. Различают следующие варианты неврологических выпадений: анестезия промежности и нижних конечностей, отсутствие бульбокавернозного, анального, ахиллова, коленного, подошвенного и кремастерного рефлексов, что свидетельствует о со-четанном передне- и заднероговом типе поражения, а также о распространённости миелодиспластического процесса. Тяжёлые трофические нарушения в виде трофических язв промежности и нижних конечностей возникают у трети больных.
Характерно отставание развития одной из нижних конечностей (или обеих), что выражается в её укорочении, уменьшении размера стопы, мышечной атрофии, чрезмерной потливости или сухости кожных покровов, цианотичности, бледности, изменении температуры кожи. Причём при двусторонней патологии эти симптомы бывают выражены неодинаково справа и слева.
Тазовые расстройства присоединяются к уже имеющимся неврологическим двигательным и чувствительным нарушениям. Однако следует иметь в виду, что нарушение функций тазовых органов (хронические запоры или слабость замыкательного аппарата прямой кишки с недержанием кала, тотальное недержание мочи, отсутствие позыва на мочеиспускание) может появиться и задолго до развития выраженной неврологической симптоматики.
Гидроцефалию встречают у 30% больных со спинномозговыми грыжами. Своевременная нейросонография головного мозга позволяет чётко верифицировать диагноз и при наличии нарастающей внутричерепной гипертензии провести вентрикуло-перитонеальное шунтирование желудочков мозга и таким образом остановить нарастание гидроцефально-гипертензионного синдрома (рис. 3-22).
Недержание мочи — ведущий симптом в клинике миелодиспла-зии, возникающий в 90% случаев. Проявления этого признака разнообразны и встречаются в виде поллакиурии (с частотой мочеиспусканий до 40—60 раз в сутки), императивного недержания мочи (позыв на мочеиспускание, как правило, отсутствует или проявляется в виде болевого эквивалента), энуреза.
Нейрогенный мочевой пузырь при миелодисплазии имеет свои особенности. Они связаны как минимум с двумя обстоятельствами. Первое — локализация порока и его отношение к сложной системе иннервации мочевого пузыря. Недоразвитие спинного мозга на уровне L —S3 сегментов, где преимущественно расположены центры мочеиспускания, определяет возможные варианты пузырных дисфункций. Второе — ребёнок рождается с нарушенной иннервацией мочевого пузыря, поэтому естественный этап формирования рефлекса на мочеиспускание выпадает.
Двигательные дисфункции мочевого пузыря усугубляются вторичными прогрессирующими расстройствами кровоснабжения детрузора, внутрипу-зырной гипертензией и хроническим воспалением. К моменту обращения больного к врачу в мочевой системе создаётся подчас крайне неблагоприятная ситуация, проявляющаяся тотальной лейкоцитурией, бактериурией, болями в животе, интоксикацией, недержанием мочи и т.д. В связи с этим не

обходимо применение большого и разнопланового комплекса клинических, рентгенологических, уродинамических и электрофизиологических методов исследования.
Серьёзная проблема в лечении больных с миелодисплазией — незаторможенный (неадаптированный, нестабильный) мочевой пузырь — характерная для этого заболевания форма нарушения регуляции акта мочеиспуска
ния. Мочевой пузырь следует называть нестабильным, если между двумя актами мочеиспускания, т.е. в фазу накопления, детрузор вызывает повышение внутрипузырного давления при воздействии любых раздражителей. По одной из точек зрения, сакральный парасимпатический центр мочевого пузыря находится в гиперактивном состоянии и получает тормозящие влияния со стороны вышележащих отделов спинного мозга. При задержке развития супраспинальных центров или пороках развития спинальных проводников гиперактивное состояние спинального центра вызывает хаотичные незаторможенные сокращения детрузора в фазу наполнения, т.е. приводит к развитию незаторможенного мочевого пузыря.
Незаторможенная активность мочевого пузыря и нарушения уродинами-ки по типу интермиттирующей гипертензии обусловлены резким возбуждением всех элементов эфферентного звена парасимпатической нервной системы, включающей сакральный центр, тазовые нервы, М-холинорецепторы, что подтверждается эффективностью блокады или пересечением сакральных корешков (ризидиотомия). Правильная интерпретация этого факта имеет огромное значение для выбора адекватной тактики хирургического лечения.
Отставание в развитии супраспинальных центров при наличии врождённой патологии каудальных отделов спинного мозга приводит к отсутствию подчинения наружного уретрального сфинктера волевому контролю. В норме рефлекторное расслабление детрузора сопровождается рефлекторным спазмом сфинктеров, а при детрузорно-сфинктерной диссинергии непроизвольное сокращение детрузора сопровождается сокращением сфинктеров. Подобная уретральная обструкция вызывает острое повышение внутрипузырного давления, превышающее микционное. Клинически это состояние проявляется поллакиурией, императивными позывами (при сохранности рефлекторной дуги), неудержанием мочи, нелокализованными болями и т.д. При рентгенологическом исследовании у подобных больных часто обнаруживают пузырно-мочеточниковые рефлюксы, вплоть до мегауретера.
Недержание кала — один из основных клинических признаков ми-елодисплазии, его наблюдают у 70% больных. Истинного недержания кала у таких детей практически не бывает в связи с явлениями хронического копростаза, а недержание заключается в виде постоянного каломазания.
Выраженные денервационные изменения со стороны нижних конечностей в виде вялого парапареза, паретической косолапости отмечают у 60% больных. Причём степень и распространённость пареза могут варьировать; он выражен тем меньше, чем каудальнее расположен дефект спинномозгового канала.
В тяжёлых случаях спинномозговая грыжа сопровождается нижним парапарезом и нарушением функций тазовых органов. Ребёнок постоянно бывает мокрым, так как кал и моча выделяются непрерыв
но, вызывая мацерацию кожи. Тонус наружного анального сфинктера отсутствует, анус часто зияет. Нижние конечности согнуты в тазобедренных суставах и расположены под прямым углом к туловищу. Все эти симптомы свидетельствуют о глубоких расстройствах иннервации и выраженном недоразвитии спинного мозга.
Диагностика
В последние годы широкое распространение получила внутриутробная пренатальная диагностика с помощью УЗИ. При выявлении тяжёлых форм спинномозговой грыжи, равно как и других тяжёлых пороков развития черепа, позвоночника, головного и спинного мозга, показано прерывание беременности.
Все варианты спинномозговой грыжи могут сочетаться с пороками развития головного или спинного мозга на другом уровне, поэтому таким больным следует выполнять рентгенографию всего позвоночника. Наряду с этим следует проводить УЗИ, КТ и МРТ, позволяющие выявить диастематомиелию, сирингомиелию, объёмные образования типа липом, фибром, тератом, ликворных кист, наличие дермального синуса, что в принципе меняет тактику лечения (рис. 3-23, 3-24, 3-25 и 3-26).
Дифференциальную диагностику проводят главным образом с тератомами крестцово-копчиковой области, для которых характерны дольчатость строения, наличие плотных включений и асимметричное расположение опухоли. Поставить правильный диагноз помогает рентгенологическое исследование, выявляющее при спинномозговой грыже незаращение дужек позвонков.
| Лечение |
Единственный правильный и радикальный метод терапии — хирургическое лечение. Оно показано сразу после установления диагноза. При небольших грыжах с хорошим кожным покровом, если отсутствуют нарушения функций тазовых органов и нижних конечностей, к решению вопроса об операции нужно подходить очень осторожно, так как в результате травматизации интимно припаянных к грыжевому мешку элементов спинного мозга после операции могут развиться неврологические нарушения ятрогенного характера.

Сущность операции состоит в удалении грыжевого мешка и пластике дефекта дужек позвонков. В период новорождённое™ показаниями к хирургическому вмешательству по поводу менингоради-кулоцеле считают разрыв оболочек грыжи и возможность инфицирования с развитием менингита. Если такой непосредственной угрозы нет, оперативное лечение следует отложить на более поздний срок (1-1,5 года) и проводить его нужно в специализированном стационаре с использованием прецизионной микрохирургической техники. Если ребёнок был прооперирован в раннем возрасте и у него сохраняются стойкие неврологические нарушения, резистентные к проводимому консервативному лечению, необходимо тщательное всестороннее обследование для решения вопроса о возможной реконструкции проводникового аппарата спинного мозга.
Изучение результатов хирургического лечения аномалий спинного мозга свидетельствует, что восстановление его временно утраченных функций происходит у части больных после устранения сдав-ления спинного мозга, т.е. после ликвидации воздействия таких постоянных раздражителей, как костные остеофиты, арахноидальные спайки и кисты, эпидуральные рубцы, липомы, липофибромы и т.д. Исходя из этого, сдавление спинного мозга (т.е. наличие очага пато
логической ирритативной импульсации), имеющееся у ряда больных с врождёнными аномалиями позвоночника и спинного мозга, необходимо устранять хирургическим путём. Правильность такого принципиального положения подтверждают ближайшие и отдалённые результаты хирургического лечения.
В основе хирургического лечения менингорадикулоцеле (или последствий ранее проведённого удаления грыжи) и других доброкачественных заболеваний каудальных отделов позвоночника и спинного мозга лежат следующие принципы.
• Ликвидация очага эфферентной патологической импульсации.
• Восстановление анатомо-топографических взаимоотношений элементов конского хвоста и попытка реиннервации нижележащих сегментов.
• Улучшение гемодинамики в зоне поражения и восстановление нормального тока ликвора.
Подобного эффекта можно добиться путём микрохирургического радикулолиза с прецизионным иссечением всех Рубцовых сращений, ликворных кист и других патологических интрарадикулярных образований.
Сложность хирургической тактики заключается в том, что с устранением спинномозговой грыжи оперативное лечение у большинства детей не заканчивается. Необходимый эффект даёт только комплексное многоэтапное лечение с привлечением специалистов разного профиля: уролога (лечение сочетанных аномалий мочевыделитель-ной системы и нарушений функций мочевого пузыря), нейрохирурга и микрохирурга (при развивающейся гидроцефалии и для проведения реиннервации тазовых органов), ортопеда (для восстановления опорной функции конечностей).
Определяя спектр лечебных мероприятий, приходится учитывать буквально все звенья гомеостаза организма ребёнка, страдающего мие-лодисплазией, ибо только такой подход может обеспечить определённый уровень клинического эффекта и социальную адаптацию ребёнка.
3.3. Врождённые кисты и свищи шеи
Врождённые кисты и свищи шеи подразделяют на боковые и срединные. Их возникновение связано с нарушением формирования этой области в эмбриональный период. Наиболее часто встречают сРединные кисты и свищи.
Срединные кисты и свищи шеи
Срединные кисты и свищи шеи — результат нарушения обратного развития щитовидно-язычкового протока. Срединный зачаток щитовидной железы, располагающийся в подъязычной области, спускается затем на шею, проходя через подъязычную кость. По пути опускания зачатка остаётся эмбриональный ход, который в норме облитерируется. В случаях полного отсутствия облитерации возникают срединные свищи, а при образовании замкнутой полости — срединные кисты шеи.
Дата добавления: 2015-09-07; просмотров: 1273;