ГЛАВА 8 2 страница
Свойства любого белка зависят от его конформации, которая в свою очередь определяется аминокислотной последовательностью. Некоторые аминокислоты в полипептидной цепи играют ключевую роль в определении специфичности, термостабильности и других свойств белка, так что замена единственного нуклеотида в гене, кодирующем белок, может привести к включению в него аминокислоты, приводящему к понижению его активности, либо, напротив, к улучшению каких-то его специфических свойств. С развитием технологии рекомбинантных ДНК появилась возможность производить специфические замены в клонированных генах и получать белки, содержащие нужные аминокислоты в заданных сайтах. Такой подход получил название направленного мутагенеза. Как правило, интересующий исследователя ген клонируют в ДНК фага М13. Одноцепочечную форму ДНК этого фага копируют с использованием олигонуклеотидного праймера, синтезированного таким образом, чтобы в ген-мишень был встроен определенный нуклеотид. Затем трансформируют двухпепочечными ДНК М13 клетки Е. coli. Часть образующихся в клетках фаговых частиц несет ген, содержащий нужную мутацию. Такие частицы идентифицируют, встраивают мутантный ген в экспрессирующий вектор, синтезируют белок и определяют его активность. Вносить изменения в клонированные гены можно также с помощью плазмид или ПЦР. Обычно заранее не известно, какую именно аминокислоту (аминокислоты) необходимо заменить для того, чтобы улучшить то или иное свойство белка-мишени. Поэтому предпочтительно использовать случайный, а не олигонуклеотид-направленный мутагенез.
Выбор аминокислоты, подлежащей замене, как правило, производится с учетом ее роли в функционировании белка. Данные об этом получают в ходе генетических исследований или методом ренттеноструктурного анализа трехмерной структуры белка. Изменяя специфические сайты или целые участки белковой молекулы, можно повысить термостабильность белка, изменить его чувствительность к pH, специфичность, аллостерическую регуляцию, потребность в кофакторе и другие свойства. Так, термостабильность триозофосфатиозомеразы удалось повысить, заменив аминокислоты в двух позициях. Этот подход можно использовать как для придания новых свойств уже существующим белкам, так и для создания уникальных ферментов.
ЛИТЕРАТУРА
Ahern Т. J., J. I. Casai, G. A. Petsko, A. M. Kli-
banov. 1987. Control of oligometric enzyme thermostability by protein engineering. Proc. Nati. Acaà. Sei. USA 84:675-679. Brange J., U. Ribel, J. F. Hansen, G. Dodson, M, T. Hansen, S. Havdund,S. G.Mdberg, F. Norris, K. Noms, L. Snel, A. R.Sorensen, H. O, Voigt.1988. Monomeric insulins obtained
176 ГЛАВА 8
by protein engineering and their medical implications. Nature 333: 679-682.
Chen K.( F. H. Arnold.1991. Enzyme engineering for nonaqueous solvents: random mutagenesis to enhance activity of subtilisin E in polar organic med'm.Bio/Technology 9: 1073-1077.
Deng W. P., J.A. Nickoloff. 1992. Site-directed mutagenesis of virtually any plasmid by eliminating a unique site. Anal. Biochem. 200:81—88.
Geisselsoder J., F. Witney, P. Yuckenberg.1987. Efficient site-directed in vitro mutagenesis. Bio Techniques Si 786-791.
Herlitze S., M. Koenen.1990. A general and rapid mutagenesis method using polymerase chain reaction. Gene91: 143-147.
Hermes J. D., S. M. Parekh, S. C. Blacklow, H. Küster, J. R. Knowles.1989. A reliable method for random mutagenesis: the generation of mutant libraries using spiked oligodeoxyri-bonucleotide primers. Gene 94: 143—151.
Keyt B. A., N. F. Paoni, C. J. Refino, L. Berleau, H. Nguyen, A, Chow, J. Lai, L. Pena, C. Pater, J. Ogez, T. Etcherary, D.Bofetein, W. F. Bennett.1994. A faster-acting and more potent form of tissue plasminogen activator. Proc. Natl. Acad. Sei. №/(91:3670-3674.
Kim Y.-G., J. Cha, S. Chandrasegaran.1996. Hybrid restriction enzymes: zinc finger fusions to Fok\ cleavage domain. Proc. Natl. Acad. Sei. USA 93: 1156-1160.
KirchhoffF., R. C. Desrosiers.1995. Random mutagenesis of short target DNA sequences via PC R with degenerate oligonucleotides. Methods Mol. ß/o/. 57: 323-333.
Landt О., H.-P. Grunert, U. Hahn.1990. A general method for rapid site-directed mutagenesis usirig the polymerase chain reaction. Седа 96: 125-128.
Mark D. F.,S. D. Lu,A. A. Creasey, R. Yaniaiiioto, L.S. Lin. 1984. Site-specific mutagenesis of the human fibroblast interferon gene. Prof. Natl. Acad. Sei. USA 81: 5662-5666.
Matsumura M., G. Signor, B. W. Mathews.1989. Substantial increase of protein stability by multiple disulfide bonds. Nature 342: 291-293.
Nosoh Y., T. Sekiguchi.1990. Protein engineering forthermostability. Trends Biotechnol. 8: 16-20.
Piccftocki M. P., R.N. Hines.1994. Oligonucleotide design and optimized protocol for site-directed mutagencsis. Bio Techniques 16: 702—707.
Sliiraishi H., Y. Shimura. 1988. A rapid and efficient method for targeted random mutagenesis. Gene 64:313-319.
Smith M.1985. In vitro mutagenesis. Annu. Rev. Genet. 19: 423-462.
Strausberg S. L., P. A. Alexander, D. T. Gallagher, G. L.Gilliliuid, B. L. Barnett, P.N. Bryan. 1995. Directed evolution of a subtilisin with calcium-independent stability, Bio/Technology 13: 669-673.
Wakarchuk W. W., W. L. Sung, R.L. Campbell, Λ. Cunningham, D. C. Watsnn, M. Yaguclii. 1994 Thermostabilization of the BadIlus circulons xylanase by the introduetion of disulfitle bonds. Protein Eng. 7: Ш9-1386.
Wetze! R., L. J. Perry, С. Veilleux.1991. Mutations in human interferon gamma affecting inclusion body formation identified by a general immuno-chemical screen. Bio/Technology 9: 731—737.
Wilkinson A. J., A. R. Fersht, D. M. Blow, P. Carter, G. Winter.1984. A large increase in enzyme-substrate affinity by protein engineering. Nature 307: 187-188.
Zoller M. J,, M. Smith.1982. Oligonucleotide-directed mutagenesis using M13-derived vectors: an efficient and general procedure for the production of point mutations in any fragment of DNA. Nucleic Acids Res. 10: 6487-6500.
КОНТРОЛЬНЫЕ ВОПРОСЫ
1. Какие физические и химические свойства ферментов можно изменить с помощью направленного мутагенеза?
2. Предположим, что вы клонировали бактериальный ген, экспрессирующийся в E. coli, и хотите изменить его активность. Однако в результате применения стандартного метода мутагенеза с использованием ДНК М13 по ряду технических причин лишь небольшая часть клонов приобрела мутантный ген-мишень, а большинство из них содержит интактный ген. Как увеличить долю клонов, содержащих ДНК с нужной мутацией?
3. Предположим, что вы выделили ген фермента, синтезирующегося в E. coli. Опишите стратегию изменения каталитической активности этого фермента, принимая во
Направленный мутагенез и генная инженерия белков 177
внимание тот факт, что вы знаете нуклеотидную последовательность кодирующего его гена, но не знаете, какой из участков молекулы фермента ответствен за каталитическую активность.
4. Каковы преимущества и недостатки олигонуклеотид-направленного мутагенеза с использованием бактериофага М13 и ПЦР?
5. Опишите стратегию олигонуклеотид-направленного мутагенеза с использованием плазмидной ДНК.
6. Как, используя «вырожденные" праймеры, можно вносить случайные мутации в ДНК?
7. Опишите стратегию повышения стабильности белка, в котором а) отсутствуют остатки цистеина; б) присутствует нечетное число остатков цистеина.
8. Как влияет замена аспарагина на другой аминокислотный остаток на стабильность белка?
9. Каким образом можно изменить потребность фермента в кофакторах?
10. Как вы будете изменять каталитическую активность или субстратную специфичность фермента, ген которого вы выделили? В чем смысл этой процедуры?
ЧАСТЬ II.
Mолекулярная
биотехнология
микробиологических
систем
С развитием технологии рекомбинантных ДНК появилась возможность более эффективно использовать многие полезные свойства микроорганизмов. В ч. II мы рассмотрим некоторые примеры практического применения микробиологических систем, модифицированных методами генной инженерии.
С помощью современных генетических методов биологи научились превращать бактерии в своеобразные «биологические фабрики» по производству белковых препаратов (например, рестрицируюших эндонуклеаз), различных химических соединений, аминокислот, антибиотиков и т. д. Клонируя в бактериальных клетках специфические гены, они создают новые пути биосинтеза для получения уникальных метаболитов, применяют клонированные гены болезнетворных микроорганизмов в качестве зондов для диагностики заболеваний человека и домашних животных, используют изолированные гены для получения безопасных и эффективных вакцин.
Методами генной инженерии можно усиливать природную способность определенных видов бактерий к осуществлению специфических биологических процессов. Например, уже получены штаммы бактерий, которые более эффективно разрушают токсичные отходы, загрязняющие окружающую среду, способствуют ускорению роста сельскохозяйственных культур, эффективно расщепляют целлюлозу до низкомолекулярных углеродных соединений, уничтожают вредных насекомых.
Часто думают, что выращивание больших количеств микроорганизмов представляет собой рутинную процедуру. Однако для успешного производства рекомбинантных белков в промышленных масштабах необходимо контролировать множество параметров, от которых зависит рост синтезирующих их микроорганизмов и чистота получаемых продуктов.
ГЛАВА 9.
Молекулярная диагностика
Успехи современной медицины и сельского хозяйства часто зависят от того, удается ли обнаруживать специфические вирусы, бактерии, грибы, паразитические микроорганизмы, белки и низкомолекулярные соединения в организме человека или животных, в растениях, воде или почве. Например, профилактику и лечение любого инфекционного заболевания значительно облегчает ранняя и точная идентификация вызвавшего его патогенного микроорганизма. Для проведения многих диагностических процедур необходимо сначала вырастить культуру потенциально патогенного микроорганизма и лишь затем проанализировать спектр его физиологических свойств. Хотя подобные тесты весьма эффективны и обладают достаточно высокой специфичностью, они часто занимают много времени и являются дорогостоящими. Это относится к идентификации и бактерий, и паразитических микроорганизмов (табл. 9,1). Кроме того, весьма ограничена возможность выявления тех патогенных микроорганизмов, которые пло-
| Таблица 9,1, Сравнение некоторых методов диагностики инфекционных заболеваний, вызванных паразитическими микроорганизмами1' | ||
| Метод | Преимущества | Недостатки |
| Mикроскопическое | Простота | Трудоемкость, длительность |
| исследование | Прямое выявление паразитических | Низкая чувствительность |
| микроорганизмов | Невозможность разграничения сходных | |
| Возможность разграничения | м икроорганизмов | |
| микроорганизмов по морфологическим | Необходимость высокой квалификации | |
| признакам | для интерпретации результатов | |
| Культивирование in vitro | Обнаружение только жизнеспособных | Длительность анализа, высокая стоимость |
| и иммунизация мышей | паразитических микроорганизмов | Разные породы животных лают разные ответы |
| Возможность определения вирулентности | Потеря жизнеспособности в организме животного | |
| и инфекционности | Использование животных | |
| Определение антител в | Простота, непродолжительность анализа | Не всегда специфичен |
| сыворотке | Возможность автоматизации | Невозможность разграничения острой и латентной |
| Возможность тестирования большого | форм инфекции | |
| числа образцов | ||
| Гибридизация и ПЦР | Быстрота, высокие чувствительность и | Высокая стоимость анализов, многоэтапность |
| специфичность | Невозможность различить живые и мертвые | |
| Прямое обнаружение паразитических | микроорганизмы | |
| м и к роорган измо в | Возможность получения лож неположительных | |
| Возможность разграничения разных видов | и ложноотрицательных результатов | |
| Независимость результатов от предыдущих | ||
| инфекций | ||
| Не требуют жизнеспособности | ||
| паразитов | ||
| Возможность автоматизации | ||
| 1) По данным работы Weiss, Clin. Microbiol. Rev. 8: 113-130, 1995. |
182 ГЛАВА 9
хо растут в культуре либо вообще не поддаются культивированию. В качестве примера можно привести облигатных внутриклеточных паразитов Chlamydia trachomatis, которые вызывают хламидиоз, болезнь, передающуюся половым путем и распространенную в Северной Америке и Европе, Хламидиоз трудно диагностировать, поскольку для этого необходима перевиваемая культура клеток. При этом часто получают ложноотрицательные результаты (т. е. ошибочно диагностируют отсутствие микроорганизма), в результате чего не проводится адекватное лечение. Безусловно, если для выявления микроорганизма необходимо выращивать его в культуре, то рутинной может стать идентификация лишь нескольких из всех известных патогенных микроорганизмов. Чтобы устранить это принципиальное ограничение, были разработаны методы молекулярной диагностики, в основе которых лежат иммунологические подходы или методы обнаружения специфической ДНК.
Любой метод выявления патогенных микроорганизмов должен быть достаточно простым и обладать высокой специфичностью и чувствительностью. Специфичный диагностический тест должен давать положительный ответ только на микроорганизм- или молекулу-мишень, чувствительный — обнаруживать очень малые количества такой мишени даже на фоне других микроорганизмов или молекул, загрязняющих образец. Простота метода подразумевает, что он является достаточно продуктивным, эффективным и недорогим для рутинного применения.
По оценкам специалистов, объем мирового рынка иммунодиагностических тестов в 1993 г. составил 3,4 млрд. долл. США и в ближайшие 10—15 лет будет возрастать на 5—10% ежегодно. В 1994г. объем мирового рынка ДНК-диагностических тестов был равен примерно 80 млн. долл., к 2000 г. он, по-видимому, составит 600 млн. долл., а к 2004 г. — 2 млрд. долл. В этой главе мы обсудим принципы некоторых методов молекулярной диагностики и сферу их применения.
Методы иммунодиагностики
Многие иммунологические системы детекции обладают высокой чувствительностью и специфичностью, являясь в то же время достаточно
простыми. Они широко используются для тестирования лекарственных препаратов, оценки и мониторинга различных онкологических заболеваний, определения специфических метаболитов, идентификации и контроля патогенных микроорганизмов, но имеют и свои ограничения. Если молекулой-мишенью является белок, то необходимо обеспечить экспрессию детерминирующих его генов и создать условия, в которых не происходит маскирование или блокирование сайта связывания с антителом.
Традиционные процедуры диагностики возбудителей инфекции опираются либо на набор характеристик патогенного микроорганизма, либо, что предпочтительнее, на одну уникальную, легко различимую его особенность. Клинические микробиологи пытаются найти тот минимальный набор биологических характеристик, при помощи которого можно будет гарантированно обнаруживать и идентифицировать патогенные микроорганизмы. Например, некоторые возбудители вырабатывают специфические биохимические соединения, которые и необходимо обнаружить в биологическом образце. Часто подобную маркерную молекулу можно выявить непосредственно, проведя высокоспецифичный биохимический анализ. Но такой подход неизбежно приведет к увеличению числа индивидуализированных систем детекции патогенных микроорганизмов. Более предпочтительным был бы универсальный метод, позволяющий выявлять любую маркерную молекулу независимо от ее химической природы. Именно таким является метод, основанный на идентификации комплексов антиген—антитело.
Ферментный иммуиосорбентный анализ
Существует целый ряд подходов, позволяющих определить, произошло ли связывние антитела с антигеном-мишенью. Один из них — это ферментный иммуносорбентный анализ (ELISA), который часто используют для диагностики. Процедура включает следующие этапы (рис. 9.1).
1. Образец, в котором хотят обнаружить специфическую молекулу или микроорганизм, фиксируют на твердой подложке, например на пластиковой микротитровальной плашке, обычно имеющей 96 лунок (рис. 9.1, А).
Молекулярная диагностика 183
2. К фиксированному образцу добавляют антитело, специфичное к маркерной молекуле (первое антитело), затем промывают лунку, чтобы удалить несвязавшиеся молекулы первого антитела (рис. 9.1, Б).
3. Добавляют второе антитело, которое специфически связывается с первым антителом и не взаимодействует с маркерной молекулой (рис. 9.1, В). К этому антителу присоединен фермент (например, щелочная фосфатаза, пероксидаза или уреаза), катализирующий превращение неокрашенного субстрата в окрашенный продукт. Промывают лунку, чтобы удалить несвязавшиеся молекулы конъюгата второе антитело—фермент.
4. Добавляют неокрашенный субстрат (рис.9.1,/).
5. Проводят качественное или количественное определение окрашенного продукта.
Если первое антитело не связывается с мишенью образца, то оно удаляется при первом промывании. Поскольку при этом конъюгату второе антитело—фермент не с чем связываться, он удаляется при втором промывании, и образец
| Рис. 9,1, Обнаружение антигена-мишени с помощью ELISA. Е— фермент, присоединенный ко второму антителу. | 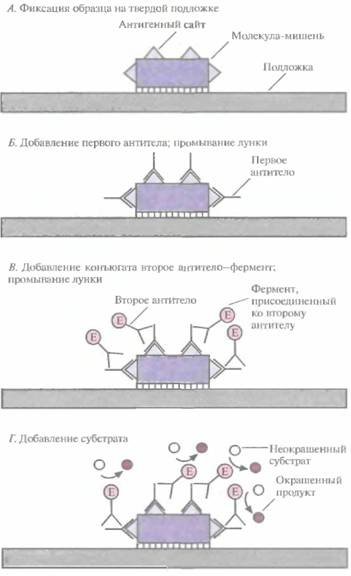
|
184 ГЛАВА 9
остается неокрашенным. Если связывание с мишенью происходит, то второе антитело присоединяется к первому, и конъюгированный фермент катализирует образование легко регистрируемого окрашенного продукта.
Основной принцип ELISA — специфическое связывание первого антитела с мишенью. Если молекула-мишень представляет собой белок, то его очищенный препарат обычно используют для получения антител, при помощи которых затем и выявляют данную мишень. Антитела, которые образуются в сыворотке (антисыворотке) крови иммунизированного животного (обычно кролика), связываются с разными антигенными детерминантами (эпитопами) молекулы-мишени. Такую смесь антител называют поликлональным препаратом. Использование поликлональных антител имеет два недостатка, существенных для некоторых методов диагностики: 1) содержание отдельных антител в поликлональном препарате может варьировать от одной партии к другой; 2) поликлональные антитела нельзя применять, если необходимо различить две сходные мишени, т. е. когда патогенная (мишень) и непатогенная (не-мишень) формы различаются единственной детерминантой. Однако эти проблемы вполне разрешимы, поскольку сейчас научились получать препараты антител, выработанных к одной антигенной детерминанте, т. е. препараты моноклональных антител.
Моноклональные антитела
У млекопитающих в ходе эволюции выработался сложный набор клеточных систем, защищающих организм от токсичных веществ и инфекционных агентов. Составной частью защитной реакции является индуцированная выработка клетками лимфатической системы специфических белков (антител), которые соединяются с чужеродными веществами (антигенами) и при помощи других белков иммунной системы, включая системы комплемента, нейтрализуют их эффект. В ответ на иммунологический стимул каждая антителопродуцирующая клетка синтезирует и выделяет единственный вид антител, которые с высоким сродством распознают отдельный участок (эпитоп, антигенную детерминанту) молекулы антигена. Поскольку в молекуле антигена обычно присутствует несколько разных эпитопов, антитела против каждого из них вырабатываются отдельными клетками иммунной системы. Такие антитела, каждое из которых взаимодействует с данным антигеном, называют поликлональными.
Уже в начале нынешнего века, когда о поликлональности антител ничего не знали, было ясно, что их специфичность можно использовать для подавления инфекций. Позже антитела стали применять в качестве диагностического инструмента для выявления токсичных соединений в клинических образцах. К сожалению, эффективность препаратов поликлональных антител варьирует от одной партии к другой, поскольку в одних случаях при проведении иммунизации антителoпродуцирующие клетки сильнее стимулируются одними детерминантами данного антигена, а в других иммунная система активнее отвечает на другие эпитопы того же антигена. Это может влиять на способность разных препаратов нейтрализовать антигены, поскольку отдельные эпитопы обладают разной эффективностью (стимулирующей способностью). Следовательно, в данной партии поликлональных антител может содержаться мало молекул, направленных против основного эпи-топа, и в результате она будет менее эффективной, чем предыдущая.
Следовательно, для практического применения антител в качестве диагностического инструмента или компонентов терапевтических средств необходимо было создать такую линию клеток, которая росла бы в культуре и продуцировала антитела одного типа, обладающие высоким сродством к специфическому антигену-мишени, — моноклональные антитела. Подобная клеточная линия могла бы стать неиссякающим источником идентичных молекул антител. К сожалению, В-лимфоциты (B-клетки), синтезирующие антитела, не могут воспроизводиться в культуре. Решение данной проблемы виделось в создании гибридной клетки. Получив генетическую составляющую от B-клетки, она могла бы вырабатывать антитела, а приобретя способность к делению от клетки совместимого типа — расти в культуре. Было известно, что В-лимфоциты иногда перерождаются и становятся раковыми (миеломными) клетками, приобретая спо-
Молекулярная диагностика 185
собность к росту в культуре и сохраняя в то же время многие свойства B-клеток. Так клетки миеломы, в первую очередь те, которые не вырабатывают антител, стали кандидатами на слияние с антителопродуцирующими В-клетками. В середине 70-х гг. эти идеи стали реальностью.
Образование и отбор гибридных клеток
Первый шаг в процессе получения гибридной клеточной линии, продуцирующей антитела одного типа, состоит во введении мышам антигена. После ряда иммунизации, проведенных в течение нескольких недель, проверяют, произошло ли развитие у животных иммунного ответа. Если ответ развился, то животных умерщвляют, извлекают селезенку, промывают ее, измельчают и несильно встряхивают для высвобождения единичных клеток, среди которых находятся и антителопродуцирующие B-клетки. Взвесь клеток селезенки смешивают со взвесью миеломных клеток, дефектных по гипоксантин-гуанин— фосфорибозилтрансферазе (HGPRT~). Комбинированную взвесь в течение нескольких минут инкубируют в 35%-ном полиэтиленгликоле, а затем переносят в среду, содержащую гипоксантин, аминоптерин и тимидин (среда ГАТ).
Обработка полиэтилен гликолем облегчает слияние клеток, тем не менее слияние происходит редко и является в достаточной степени случайным событием. В смеси присутствуют клетки миеломы, селезенки, а также слившиеся клетки миеломы-селезенки, миеломы-миеломы, селезенки—селезенки. Однако в среде ГАТ растут только гибридные клетки миеломы—селезенки, все остальные типы клеток не могут в ней пролиферировать. Клетки селезенки и слившиеся клетки селезенки—селезенки вообще не растут в культуре, а миеломные клетки HGPRT~ и слившиеся клетки миеломы—миеломы не могут использовать гипоксантин в качестве предшественника в процессе биосинтеза пуриновых оснований гуанина и аденина, без которых невозможен синтез нуклеиновых кислот. Но у них есть другой естественный путь синтеза пуринов — при участии дигидрофолатредуктазы, поэтому в состав среды и входит аминоптерин, ингибирующий активность этого фермента. Таким образом, миеломные клетки HGPRT~ и слившиеся клетки миеломы-миеломы не могут синтезировать пурины в среде ГАТ и погибают.
Слившиеся клетки селезенки—миеломы растут в среде ГАТ, поскольку; 1 ) клетки селезенки поставляют функциональную HGPRT, которая может утилизировать экзогенный гипоксантин среды несмотря на блокирование синтеза пуринов с участием дигидрофолатредуктазы аминоптерином; 2) клетки миеломы способны активно делиться. Тимидин необходим для устранения блокирования в синтезе пиримидинов, обусловленного ингибированием дигидрофолатредуктазы. На 10-14-е сутки после слияния клеток вереде ГАТ остаются и растут только слившиеся клетки селезенки—миеломы. Их затем вносят в лунки пластиковых микротитровальных плашек и выращивают на полной культуральной среде без ГАТ.
Идентификация гибридных клеточных линий, секретирующих специфические антитела
Теперь необходимо идентифицировать гибридные клетки, вырабатывающие антитела к иммунизирующему антигену. Для этого обычно проводят скрининг культуральных сред, содержащих секретируемые антитела. Среду из тех лунок, в которых есть растущие клетки, отбирают и переносят в лунки другой микротитровальной плашки, предварительно покрытые слоем молекул антигена-мишени. Если в культуральной среде находится антитело (первое антитело), распознающее один из эпитопов данного антигена, то оно свяжется с антигеном и останется в лунках после их промывания. Затем в лунки добавляют второе антитело, специфичное к мышиным антителам. Оно будет присоединяться к любому первому антителу, связанному с антигеном.
К используемому в иммунном анализе второму антителу предварительно присоединяют фермент, который превращает неокрашенный субстрат в окрашенное соединение. Изменение цвета содержимого одной из лунок говорит о том, что исходная культуральная среда содержала антитело, специфичное к данному антигену (рис. 9.2). Если же такое антитело в среде отсутствовало, то второму антителу не с чем будет
186 ГЛАВА 9
связываться и оно смоется при втором промывании. Субстрат в таких лунках останется неокрашенным.
Лунки исходной микротитровальной плашки, среда из которых дает положительный иммунный ответ (изменение цвета), могут содержать смесь слившихся клеток. Чтобы получить линии, происходящие от одной клетки (клоны), клеточную суспензию из таких лунок разводят культуральной средой и высевают в другие лун-
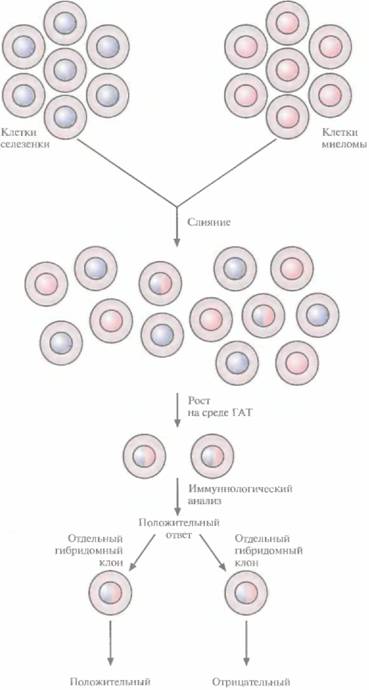
| Рис. 9.2. Скрининг клеток, вырабатывающих моноклональные антитела. Выделяют клетки селезенки мыши, иммунизированной специфическим антигеном, и проводят их слияние с клетками миеломы, не вырабатывающими антитела. Слившиеся клетки отбирают по способности к росту на среде ГАТ (гипоксантин, аминоптерин, тимидин). Клетки, вырабатывающие специфические антитела к иммунизирующему антигену (клетки гибридомы), идентифицируют иммунологическими методами и субкультивируют, чтобы получить отдельные клоны. Из гибридомы, растущей в культуре и секретирующей единственный тип молекул антител, получают моноклональные антитела. |
Молекулярная диагностика 187
ки. После культивирования полученных клонов среды вновь тестируют, определяя, какая из клеточных линий (гибридом) продуцирует моноклональные антитела, распознающие антиген-мишень. В том случае, когда получают более одной специфичной гибридомы, проводят дальнейшие исследования, позволяющие определить, направлены ли антитела, вырабатываемые разными клонами, против одной и той же антигенной детерминанты. Каждый клон, продуцирующий моноклональное антитело, можно поддерживать в культуре практически бесконечно. Кроме того, образцы можно заморозить в жидком азоте и использовать их в дальнейшем как источник клеток.
Дата добавления: 2015-07-14; просмотров: 947;