Cockett M. I., С. R. Beddington, G. T. Yarranton.
1990. High level expression of tissue inhibitor or metalloproteinases in Chinese hamster ovary cells using glutamine synthetase gene amplification. Bio/Technology 8: 662-667.
Cole E.S., K. Lee, K.Lau/iere, C. Kelton, S. Chappel, B. Weintraub,I). Ferrara, F. Peterson, R. Bernasconi, T. Edmunds, S. Richards,L. Dickrell, I. M. Kleeman, J. II. McPhersun, B. M. Pratt. 19УЗ. Recombinanl human thyroid stimulating hormone: development of a biotechnology product for detection of metasla tic lesions of thyroid carcinoma. Bio/Technology 11:1014-1024.
Cregg J. M., J. F. Tschopp, C. Stillman, R.Siegel, M. Akong, W.S. Craig, R. G.Buckhnl/, К. R. Madden, P. A. Kellaris, G. R. Davis,B. L. Smiley, J. Cruzc, R. Torregrossa, G. Veiicclebi, G. P. Thill.1987. High-level expression and efficient assembly of hepatitis В surface antigen in the methylotrophic yeast, Pichla pastoris. Bio/Technology 5: 479-485.
DavîesA. H.1994, Current methods for manipulating baculovirus, Bio/Technology 12:47—50.
Digan M. E.,S. V. Lair, R. A. Brieriey, R. S. Siegel, M. E. Williams, S. B. Ellis, P. A. Kellaris, S. A. Provow, W.S. Craig, G. Velicelebi, M. M. Harpold, G. P. ThiU.1989. Continuous production of a novel lysozyme via secretion from the yeast, Pichia pastoris. Bio/Technology 7: 160-164.
Dirks W., M. Wirth, H. Hauser.1993. Dicistronic transcription units lor gent expression in mammalian cells. Gene 128: 247-24'λ
Gellissen G., Z. A. Janowicz, U. Weydemann, K. Melber, A. W. M. Strasser, C. P.Hollenberg. 1992, High-level expression of foreign genes in
156 ГЛАВА 7
Hansenula polymorpha. Bioiechnol. Adv. 10: 179-189,
Giga-Hama Y., H. Tohda, H. Okada, M. K. Owada, H. Okayama, II. Kumagai. 1994. High-level expression of human lipocortin I in the fission yeast Schizosaccharomyces pombe using a novel expression vector. Bio/Technology 12: 400-404.
GilbertS. C., H.van Urk, A. J.Greenfield, M. J. McAvoy, K. A. Denton, D.Coghlan, G. D. Jones, D. J. Mead.1994. Increase in copy number of an integrated vector during continuous culture of Hansenula polymorpha expressing functional human hemoglobin. Yeast 10: 1569-1580.
The GlobalUse ofStrategies to OpenOccluded Arteries (GUSTO) lib Investigators.1996. A comparison of recombinant hirudin with heparin for the treatment of acute coronary syndromes. N. Engl. J. Med. 335: 775-782.
Hallewell R.Α., R. Mills, P. Tekamp-OLson, R. Blacher, S. Rosenberg, F. Otting, F. R. Masiarz,
C. J. Scandella.1987. Amino terminal acetyla-tion of authentic human Cu, Zn Superoxide dis-mutase produced in yeast. Bio/Technology 5: 363-366.
Kidd L M., V. C. Emery.1993. The use of baculoviruses as expression vectors. Appl. Biochem. Biotechnol. 42: 137-159.
Kitts P. A., R. D. Possee.1993. A method for producing recombinant baculovirus expression vectors at high frequency. BioTechniques 14: 810-817.
Laroche Y., V. Strome, J. DeMeutter, J.Messens, M. Lauwereys.1994. High-level secretion and very efficient isotopic labeling of tick anticoagulant peptide (TAP) expressed in the methylotrophic yeast, Picftia postons. Bio/Technology 12: 1119-1124.
Loison G., A. Findeli, S. Bernard,M. Nguyen-Juilleret, M.Marquet, N. Riehl-Bellon,
D. Carvallo, L. Guerra-Santos, S. W. Brown, M.Courtney, C. Roitsch, Y. Lemoine.1988. Expression and secretion in S. cerevisiae of biologically active leech hirudin. Bio/Technology 6: 72-77.
Lucas B. K., L. M. Giere, R, A. DeMarco, A. Shen, V.Chisholm, C. W. Crowley.1996. High-level production of recombinant proteins in CHO cells
using a dicistronic DHFR intron expression vector. Nucleic Acids Res. 24: 1774-1779.
Lucknow V. A., S. C. L«e, G. F. Barry, P. O. Olins.1993, Efficient generation of infectious recombinant baculoviruses by site-specific transposon-mediated insertioin of foreign genes into a baculovirus genome propagated in Eschericia coli. J. VIrol. 67: 4566-4579.
Peng S., M. SommtrFelt, J. Logan, Z. Huang,T. Jilling, K. Kirk, E. Hunter, E.Sorscher. 1993. One-step affinity isolation of recombinant protein using the baculovirus/insect expression system. Protein Expr. Purif. 4: 95-100.
Robinson A. S., V. Hines, K. D. Wittrup.1994. Protein disulfide isomerase overexpression increases secretion of foreign proteins in Saccharomyces cerevisiae. Bio/Technology 12:381-384,
Romanes Μ. Α., C. A. Scorer, J. J. Clare.1992, Foreign gene expression in yeast: a review. Yeast 8: 423-488.
Vozza L. A., L. Wittmer, D. R. Higgins, T. J. Purcell, M. Bergseid, L. A. Collins-Racie, E. R. LaVaUie, J. P. Hoeffler.1996, Production of a recombi-rtant bovine enterokinase catalytic subunit in the methylotrophic yeast, Pichia pastoris. Bio/ Technology 14: 77-81.
КОНТРОЛЬНЫЕ ВОПРОСЫ
1. Почему для получения белков, использующихся в медицине, лучше применять эукариотические, а не прокариотические системы?
2. Какие достоинства и недостатки имеют различные типы дрожжевых векторов, предназначенных для получения данного рекомбинантного продукта?
3. Опишите основные свойства интегративной векторной системы P. pastoris, обладающей высоким уровнем экспрессии,
4. Что такое бакуловирусы? Опишите исходную систему экспрессии на основе ба-куловирусов и ее последующие модификации.
5. Что такое бакмида? Для чего ее используют?
Получение рекомбинантных белков с помощью эукариотических систем 157
6. Что такое аффинная метка? Для чего ее используют?
7. Опишите основные свойства внехромосомного экспрессирующего вектора млекопитающих.
8. Опишите как минимум две селективные системы, использующиеся в случае экспрессирующих векторов млекопитающих.
9. Опишите разные подходы к созданию систем синтеза двух рекомбинантных белков в одной клетке млекопитающего.
10. Какими критериями руководствуются при выборе системы экспрессии генов гетерологичных белков (дрожжи, система экспрессии на основе бакуловирусов, клетки млекопитающих)?
ГЛАВА 8
Направленный мутагенез и генная инженерия белков
Технология рекомбинантных ДНК позволяет выделять гены любых белков, существующих в природе, экспрессировать их в специфическом хозяйском организме и получать чистые белковые продукты. Однако физические и химические свойства таких «природных» белков часто не удовлетворяют условиям, обеспечивающим возможность их промышленного применения. Иногда для получения белков, обладающих нужными свойствами, в качестве источника соответствующих генов используют организмы, растущие в необычных, зачастую экстремальных условиях. Например, для синтеза α -амилазы, не утрачивающей своей активности при высокой температуре, выделили ее ген из Bacillus stearothermophilus — бактерии, естественной средой обитания которой являются горячие источники с температурой воды 90 °С. Полученная таким образом α-амилаза оставалась активной при температурах, при которых осуществляют промышленное производство этилового спирта из крахмала. Для получения белков с заранее заданными свойствами можно использовать также мутантные формы генов. Однако число мутантных белков, образующихся в результате замены отдельных нуклеотидов в структурном гене с помощью обычного мутагенеза, чрезвычайно велико. Мутагенез с последующим отбором редко приводит к существенному улучшению свойств исходного белка, поскольку большинство аминокислотных замен сопровождается снижением активности фермента.
Для создания белков со специфическими свойствами можно использовать другой подход, основанный на внесении изменений в кодирующие их клонированные гены. Это позволяет получать белки с другими, чем у их аналогов, свойствами.
• Изменив константу Михаэлиса (KМ), которая характеризует прочность связывания субстрата с ферментом, и максимальную скорость (Vmax) превращения субстрата в продукт при определенных условиях, можно повысить общую каталитическую эффективность (Vmax./KM) реакции; Vmax равна полному количеству фермента (Е0 ), умноженную на каталическую константу (kcat).
• Повысив стабильность белка в широком диапазоне температур или pH, можно использовать его в условиях, при которых исходный белок инактивируется.
• Создав белки, способные функционировать в безводных растворителях, можно осуществлять каталитические реакции в нефизиологических условиях.
• Изменив белок таким образом, чтобы он мог работать без кофактора, можно использовать его в некоторых непрерывных промышленных процессах.
• Изменив активный центр фермента, можно повысить его специфичность и уменьшить число нежелательных побочных реакций,
• Повысив устойчивость белка к клеточным протеазам, можно упростить процедуру его очистки и повысить выход продукта.
• Изменив аллостерическую регуляцию фермента, можно уменьшить степень его ингиби-рования метаболитом по типу отрицательной обратной связи и увеличить выход продукта.
Направленный мутагенез и генная инженерия белков 159
Направленный мутагенез: методика
Получить новый белок с заранее заданными свойствами — непростая задача, но вполне реально изменить свойства уже существующего белка. Изменения можно вносить в сам белок или в его ген. Однако химическая модификация белков редко бывает строго специфичной и ее необходимо осуществлять заново для каждого белкового препарата, поэтому лучше вносить изменения в его клонированный ген. К сожалению, не всегда бывает известно, какую именно аминокислоту или последовательность аминокислот нужно изменить, чтобы получить белок с нужными физическими, кинетическими или химическими свойствами. Может случиться, что изменения должны затрагивать два или более аминокислотных остатка, расположенных далеко друг от друга в полипептидной цепи, но сближающихся в результате укладки белковой молекулы. Есть надежда, что уже в недалеком будущем с помощью компьютеров удастся предсказывать свойства того или иного белка, исходя из данных о его аминокислотной последовательности. Это значительно упростит процедуру создания нужных белков. Введение новой генетической информации в клонированные гены сейчас не составляет особого труда, однако чтобы определить, обладает ли искомый белок нужными свойствами, необходимо проанализировать множество белковых продуктов.
Внесение специфических изменений в коди-руюшие последовательности ДНК, приводящих к определенным изменениям в аминокислотных последовательностях, называется направленным мутагенезом. Идентификация аминокислот, замена которых даст желаемый результат, облегчается, если детально известна пространственная структура белка (ее устанавливают с помощью рентгеноструктурного анализа или других аналитических методов). Однако для большинства белков такие данные отсутствуют, поэтому направленный мутагенез — это в значительной мере эмпирическая процедура, основанная на методе проб и ошибок. Каждый белок, кодируемый мутантным геном, нужно протестировать и убедиться в том, что мутация дала желаемый эффект.
Для направленного мутагенеза клонированных генов используют разные экспериментальные подходы, В одних случаях вносят изменения в специфические сайты клонированного гена, в других случайным образом изменяют короткий фрагмент клонированного гена и среди образующихся мутантных белков выбирают один, обладающий необходимой активностью.
Олигонуклеотид-направленный мутагенез с использованием ДНК фага M13
Олигонуклеотид-направленный (сайт-специфический) мутагенез — это один из наиболее простых методов внесения точковых мутаций в клонированный ген (рис. 8.1). Для его осуществления необходимо знать: 1) точную нуклеотидную последовательность той области ДНК, которая соответствует мРНК-кодону, подлежащему изменению; 2) характер аминокислотных замен. Обычно встраивают ген-мишень в двухцепочечную форму вектора на основе бактериофага M13. Сначала выделяют одноцепочечную форму вектора (плюс-цепь M13) и смешивают ее с синтетическим олигонуклеотидом, в точности комплементарным — за исключением одного нуклеотида -- нужному сегменту клонированного гена. Этот отличающийся (т. е. неспаривающийся) нуклеотид соответствует тому нуклеотиду кодона мРНК, который необходимо изменить. В случае, представленном на рис. 8.1, триплет АТТ, соответствующий изолейциновому кодону AUU, нужно заменить на триплет СТТ, соответствующий лейциновому кодону CUU. Олигонуклеотид будет гибридизоваться с комплементарным участком клонированного гена в том случае, если: 1) он добавлен в количестве, во много раз превышающем количество ДНК М13; 2) неспаривающийся нуклеотид находится примерно посередине олигонуклеотида; 3) отжиг проводят при низкой температуре и высокой ионной силе. 3'-конец спарившегося олигонуклеотида служит затравкой для инициации синтеза ДНК, а интактная цепь ДНК M13 — матрицей. Репликация осуществляется с помощью фрагмента Кленова ДНК-полимеразы I Escherichia coli при наличии в среде четырех дезоксирибонуклеозидтрифосфатов« а присоединение последнего нуклеотида синтезированной цепи к 5'-концу затравки обеспечивает ДНК-лигаза фага Т4. Однако in vitro синтез ДНК редко идет до конца, и частично двухцепочечные молекулы приходится отделять от нормальных центрифугированием в градиенте сахарозы.
Полностью двухцепочечными молекулами ДНКфага М13, содержащими, однако, некомплементарные нуклеотиды, трансформируют клетки Е. coll. В последних образуются фаговые
160 ГЛАВА 8

| Рис. 8.1. Олигонуклеотид-направленный мутагенез. Одноцепочечную ДНК фага М13 (плюс-цепь), несущую ген-мишень, отжигают с комплементарным синтетическим олигонуклеотидом, содержащим одно основание, не комплементарное соответствующему основанию исходной ДНК, Олигонуклеотид служит затравкой для синтеза ДНК, а М13-вектор с встроенным геном — матрицей. Репликацию катализирует фрагмент Кленова ДНК-полимеразы E E. coli. Синтезированную полноразмерную цепь замыкает в кольцо ДНК-лигаза Т4. Образовавшимися двух цепочечными молекулами трансформируют E. coli. Часть фаговых частиц содержит ДНК дикого типа, часть — мутантную ДНК. |
частицы, что в конечном счете приводит к лизису клеток и образованию бляшек. Поскольку репликация идет по полуконсервативному механизму, половина популяции образующихся фаговых частиц должна содержать ДНК дикого типа, а половина — мутантную ДНК со специфической нуклеотидной заменой. Частицы, содержащие только мутантный ген, идентифицируют при помощи ДНК-гибридизации в жестких условиях, используя в качестве зонда исходный олигонуклеотид. Мутантный ген вырезают и встраивают в какой-либо экспрессирующий Е. соli-вектор. Мутантный белок синтезируют в E. coli и очищают.
На самом деле число фаговых частиц, несущих мутантную ДНК, оказывается гораздо меньше ожидаемых 50%: лишь 1—5% бляшек содержат фаг с мутантным геном. Чтобы повысить выход мутантного фага, метод олигонуклеотиднаправленного мутагенеза модифицировали. Один из подходов состоял во введении М13-вектора, несущего ген, в который необходимо внести мутацию, в штамм E. coli, дефектный по двум ферментам метаболизма ДНК (рис. 8,2). Один фермент — это мутантная форма dUTP-пирофосфатазы (dut). Клетки с неактивной dUTP-пирофосфатазой характеризуются повышенным содержанием dUTP, что приводит к
Направленный мутагенез и генная инженерия белков 161
встраиванию в ДНК при репликации нескольких остатков dUTP вместо dTTP. Второй фермент – это дефектная урацил-К-гликозилаза (ung). В отсутствие функциональной урацил-М-гликозилазы остатки dUТР, случайно встроившиеся в ДНК, не могут быть удалены. В одноцепочечной ДНК M13, синтезированной в таких клетках Е. соli, примерно 1% тимидиновых остатков оказываются замененными уридиновыми. Олигонуклеотид с некомплементарным основанием отжигают с урацилсодержащей ДНК М13 и in vitro достраивают вторую
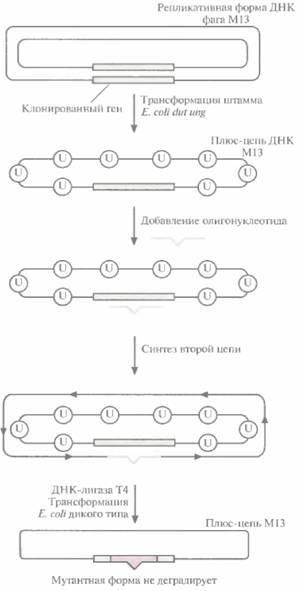
| Рис. 8.2. Повышение выхода мутантного фага M13 путем трансформации штамма Е. colt dia ung. Ген-мишень встраивают в двух цепочечную репликативную форму ДНК фага MI3 и полученными молекулами трансформируют штамм Е. coli dut ung. Мутация dut вызывает повышение содержания dUTP в клетке, что приводит к включению в ДНК нескольких остатков dUTP (U). а мутация ung блокирует их удаление. Двухцепочечной ДНК М13, содержащей ген-мишень, трансформируют клетки E. coli дикого типа. Продукт гена ung дикого типа (урацил-N-гликозилаза) удаляет все остатки урацила из исходной цепи, и она деградирует. Мутантная цепь остается интактной, поскольку она не содержит остатков урацила. Эта цепь служит матрицей для репликации ДНК, и в результате доля фаговых частиц, несущих мутантный ген, увеличивается. |
цепь. Двухцепочечной ДНК трансформируют штамм E. coli, содержащий функциональный ген ung. Активная урацил-N-гликозилаза хозяйских клеток удаляет остатки уридина из ДНК M13 (рис. 8.2), исходная матричная цепь М13 деградирует и далее реплицируется только мутантная цепь, не содержащая dUTP, В результате выход фаговых частиц, несущих мутантный ген, значительно увеличивается.
Олигонуклеотид-направлепный мутагенез с использованием плазмидной ДНК
Основной недостаток одигонуклеотид-направленного мутагенеза с использованием фага М13 — большое число процедур. Чтобы выделить мутантную форму нужного гена, приходится затратить много времени. В качестве альтернативы системе с использованием фага M13 было разработано множество других подходов, основанных на применении плазмидных ДНК. Это позволяет обойтись без переноса интересующего исследователя гена из плазмиды в фаговую ДНК, а после завершения мутагенеза — обратно в плазмиду. Один из этих подходов включает встраивание ДНК в плазмидный вектор, который несет функциональный ген устойчивости к тетрациклину и неактивный ген устойчивости к ампициллину; в середине последнего заменен один нуклеотид (рис. 8.3). Клетки E. coli трансформируют вектором, несущим ДНК-мишень, и двухцепочечную плазмидную ДНК денатурируют щелочью с тем, чтобы получить одноцепочечные кольцевые молекулы. Денатурированную ДНК отжигают с тремя разными олигонуклеоти-
162 ГЛАВА 8]
дами. Один из них предназначен для внесения изменений в клонированную ДНК-мишень, второй — для устранения мутации в гене устойчивости к ампициллину, третий — для замены одного нуклеотида в гене устойчивости к тетрациклину с тем, чтобы инактивировать этот ген. В реакционную смесь добавляют четыре дезоксирибонуклеозидтрифосфата и ДНК-полимеразу Т4, функционирующую аналогично фрагменту Кле-нова ДHК-полимеразы I Е. coli. Гибридизовавшиеся олигонуклеотиды служат затравками для синтеза ДНК, а интактная кольцевая молекула
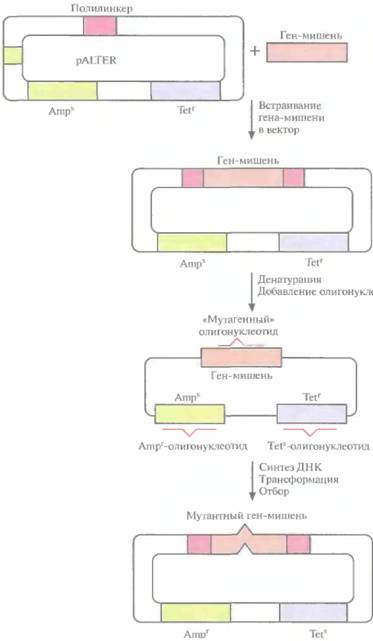
|  Рис, 8.3. Олигонуклеотид-направленный мутагенез с использованием плазмидной ДНК. Ген-мишень встраивают в полилинкер вектора pALTER. Плазмидную ДНК денатурируют в щелочи и отжигают с тремя олигонуклеотидами: «мутагенным» олигонуклеотидом, олигонуклеотидом, восстанавливающим устойчивость к ампициллину (Ampr), и олигонуклеотидом, придающим чувствительность к тетрациклину (Tets). Эти олигонуклеотиды служат затравками для синтеза ДНК с помощью ДНК-полимеразы Т4, а исходная цепь — матрицей. Одноцепочечные разрывы в новосинтезированной цепи зашиваются ДНК-лигазой Т4. Продуктами реакции трансформируют клетки E. coli и отбирают трансформантов Аmрг и TetS.
Рис, 8.3. Олигонуклеотид-направленный мутагенез с использованием плазмидной ДНК. Ген-мишень встраивают в полилинкер вектора pALTER. Плазмидную ДНК денатурируют в щелочи и отжигают с тремя олигонуклеотидами: «мутагенным» олигонуклеотидом, олигонуклеотидом, восстанавливающим устойчивость к ампициллину (Ampr), и олигонуклеотидом, придающим чувствительность к тетрациклину (Tets). Эти олигонуклеотиды служат затравками для синтеза ДНК с помощью ДНК-полимеразы Т4, а исходная цепь — матрицей. Одноцепочечные разрывы в новосинтезированной цепи зашиваются ДНК-лигазой Т4. Продуктами реакции трансформируют клетки E. coli и отбирают трансформантов Аmрг и TetS.
|
Направленный мутагенез и генная инженерия белков 163
ДНК — матрицей. Одноцепочечные разрывы в новосинтезированной цепи зашиваются с помощью ДНК-лигазы Т4. По окончании синтеза и лигирования продуктами реакции трансформируют клетки E. coli. Трансформантов отбирают по признаку устойчивости к ампициллину и чувствительности к тетрациклину. Примерно 90% из них содержат специфическую мутацию в клонированном гене. У остальных трансформантов клонированный ген не был изменен либо потому, что олигонуклеотид не гибридизовался с ним, либо потому, что он вытеснялся в ходе синтеза ДНК. Клетки, несущие мутантный клонированный ген, идентифицируют с помощью гибридизации. Все плазмиды, штаммы, ферменты, олигонуклеотиды (кроме того, который предназначен для изменения клонированного гена), а также буферы продаются в наборе, что облегчает работу.
Олигонуклеотид-направленный мутагенез с использованием ПЦР-амплификации
Более простой и быстрый метод получения больших количеств мутантных генов, альтернативный системе с использованием фага М13, -сайт-специфический мутагенез в сочетании с полимеразной цепной реакцией (ПЦР), Один из вариантов этого подхода состоит в следующем. Ген-мишень встраивают в плазмидный вектор (рис. 8.4) и помешают препарат в две пробирки. В каждую из них добавляют по два специфических праймера для ПЦР: 1 и 2 в одну пробирку, 3 и 4 — в другую. Праймеры 2 и 3 полностью комплементарны одному из участков клонированного гена или прилегающей к нему последовательности, а 1 и 3 комплементарны другому участку, но содержат один некомплементарный нуклеотид и гибридизуются с разными цепями, так что в результате происходит замена обоих нуклеотидов данной пары. Положение сайтов гибридизации праймеров l и 2 в одной пробирке и 3 и 4 — в другой таково, что ΠЦР-продукты в разных пробирках имеют разные концы. По окончании ПЦР содержимое пробирок объединяют и проводят денатурацию, а затем ренатура-цию. Поскольку концы амплифицированных молекул ДНК из двух пробирок неодинаковы, одноцепочечные ДНК из разных пробирок ассоциируют с образованием кольцевых молекул с двумя одноцепочечными разрывами. Эти разрывы репарируются in vivo после трансформации E. coli. При ренатурации одиночных цепей из одной пробирки образуются линейные молекулы. В клетках E. coti стабильно поддерживаются в виде плазмид и наследуются только кольцевые, а не линейные молекулы, при этом все они несут сайт-специфическую мутацию. Таким образом, с помощью описанного метода можно вносить точковые мутации в клонированный ген, при этом отпадает необходимость во встраивании гена в ДНК фага M13, использовании мутантных штаммов Е. coli типа dut ung и в переносе мутантного гена из М13-вектора в экспрессирующий вектор.
Случайный мутагенез с использованием «вырожденных» олигонукмотидных праймеров
К сожалению, обычно бывает неизвестно, какую нуклеотидную замену в клонированном гене нужно произвести, чтобы получить белок с нужными свойствами. Поэтому часто приходится изменять один определенный нуклеотидный сайт всеми возможными способами. Например, можно синтезировать олигонуклеотидные праймеры, в одном из сайтов которых находятся разные нуклеотиды. Такие «вырожденные» олигонуклеотиды обычно получают, добавляя в автоматический синтезатор ДНК на определенном этапе, когда к цепи должен просоединяться специфический нуклеотид, небольшое количество (до нескольких процентов) трех других нуклеотидов (рис. 8.5). В результате получается гетерогенный по одному сайту набор олигонуклеотидных праймеров, с помощью которых можно получить соответствующий набор мутантных генов-мишеней с нуклеотидными заменами в специфическом сайте.
Этот подход имеет два преимущества: 1) не нужно в точности знать, какую роль играет тот или иной аминокислотный остаток в функционировании белка: 2) поскольку в данном сайте происходят разные аминокислотные замены, могут случайно синтезироваться белки с разнообразными интересными и полезными свойствами. Конечно, если ни один из образующихся белков не обладает нужными свойствами, приходится все начинать сначала, синтезировав новый набор "вырожденных" праймеров, комплементарных другой области гена.
Дата добавления: 2015-07-14; просмотров: 1028;