Химические свойства моносахаридов
Моносахариды, являясь полигетерофункциональными соединениями, проявляют свойства как многоатомных спиртов, так и свойства карбонильных соединений – альдегидов или кетонов. Как карбонильные соединения, кетозы и альдозы в виде открытых форм вступают в реакции присоединения, а альдозы легко подвергаются окислению. Альдозы легко вступают в качественные реакции альдегидов – реакции с аммиачным раствором оксида серебра (I) и гидроксидом меди (II), окисляясь до соответствующих кислот, которые в щелочной среде неустойчивы и подвергаются деструкции углеродного скелета.
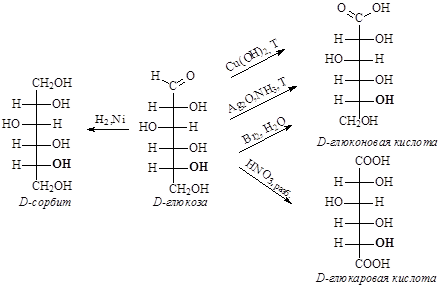
Устойчивый продукт образуется при окислении альдоз бромной водой. Следует помнить, что фруктоза в щелочной среде подвергается изомеризации в альдозы – глюкозу и маннозу, и поэтому ее растворы нельзя отличить от растворов альдоз с помощью реагентов Толленса и Фелинга, однако растворы альдоз можно легко отличить от раствора фруктозы с помощью бромной воды.
Как многоатомные спирты, моносахариды реагируют в виде циклических форм, которые преобладают в смеси таутомеров, образуя простые и сложные эфиры, ацетали.
При взаимодействии циклических полуацеталей со спиртами в кислой среде в реакцию вступает только полуацетальная гидроксильная группа, в результате реакции образуются два эпимерных ацеталя, называемых в химии углеводов гликозидами.
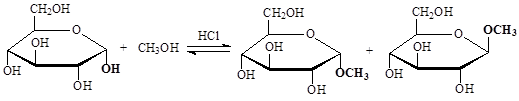
Образование двух стереоизомеров объясняется механизмом протекания реакции: под действием кислотного катализатора на первом этапе идет протонирование атома кислорода полуацетальной ОН-группы, что приводит к ее превращению в хорошо уходящую группу; на втором этапе идет отщепление молекулы воды и образование карбкатиона; на третьем этапе sp2-гибридизированный атом углерода карбкатиона подвергается нуклеофильной атаке, которая происходит с обеих сторон плоскости, что приводит к образованию стереоизомеров; на четвертом этапе происходит отщепление протона – возврат катализатора.
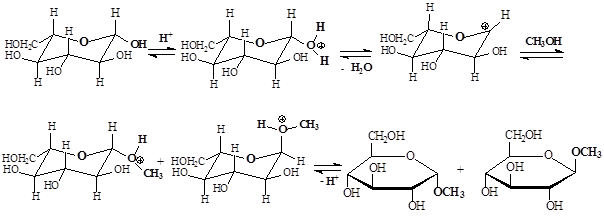
Сложные эфиры получают при действии на моносахариды ацилирующих агентов. Хотя гидроксильные группы моносахаридов несколько отличаются по реакционной способности (наиболее легко ацетилируются первичноспиртовая группа и гидроксильная группа при С2), случаи образования частично ацетилированных сахаров сравнительно редки. Ацетилирование моносахаридов со свободной полуацетальной группой осложняется наличием таутомерных превращений. При взаимодействии глюкозы с уксусным ангидридом в пиридине без нагревания образуется полный ацетат с сохранением конфигурации у С1:
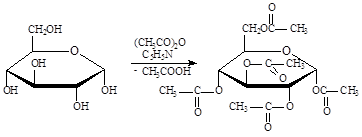
При проведении этой реакции при повышенной температуре равновесие в растворе между аномерными формами исходного моносахарида устанавливается очень быстро, и поскольку экваториальный полуацетальный гидроксил b-аномера ацетилируется значительно быстрее аксиального полуацетального гидроксила a-аномера, конечным продуктом реакции, независимо от исходной конфигурации, будет полный ацетат b-аномера:
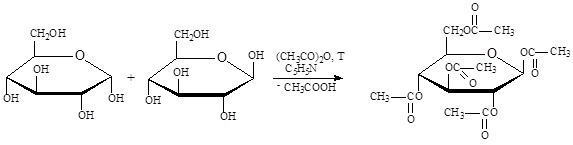
Большинство углеводов, встречающихся в природе, состоят из двух и более остатков моносахаридов, т.е. являются олиго- и полисахаридами. Формально образование этих веществ можно представить как превращение полуацеталя в ацеталь (гликозид) за счет взаимодействия полуацетальной гидроксильной группы одного моносахарида со спиртовой гидроксильной группой (реже с полуацетальной гидроксильной группой) другой молекулы моносахарида. Существенное различие биологических свойств различных олиго- и полисахаридов обусловлено особенностями пространственного строения гликозидных связей. Среди дисахаридов, являющихся простейшими представителями олигосахаридов, различают восстанавливающие и невосстанавливающие. Наиболее известными представителями восстанавливающих дисахаридов являются мальтоза, целлобиоза и галактоза.
Мальтоза (солодовый сахар) – дисахарид, получающийся при ферментативном расщеплении крахмала, состоит из двух остатков a-D-глюкопиранозы. Оба остатка глюкозы имеют пиранозный цикл и соединяются друг с другом посредством a-1,4- гликозидной связи, в образовании которой участвует полуацетальная гидроксильная группа одной молекулы и гидроксильная группа у четвертого атома углерода другой молекулы :
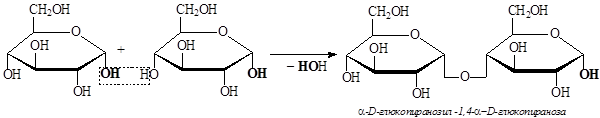
Поскольку в молекуле мальтозы содержится свободная полуацетальная гидроксильная группа, то для растворов мальтозы характерна мутаротация, обусловленная цикло-оксо-таутомерией:
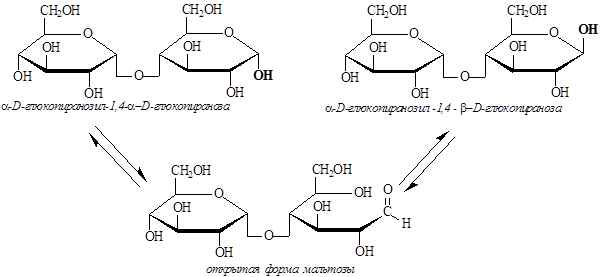
Наличие в растворах мальтозы открытой формы обуславливает проявление ею химических свойств альдегидов: она способна вступать как в реакции присоединения по двойной связи С=О (присоединять НСN, подвергаться гидрированию), так и в реакции окисления (под действием мягких окислителей она превращается в мальтобионовую кислоту). Реакции с участием гидроксильных групп приводят к образованию простых и сложных эфиров, а во взаимодействии со спиртами (в кислой среде) участвует только полуацетальная гидроксильная группа, что приводит к образованию ацеталей (гликозидов).
Целлобиоза – дисахарид, получаемый при химическом расщеплении целлюлозы, – состоит из двух остатков b-D-глюкопиранозы, соединенных b-1,4-гликозидной связью.
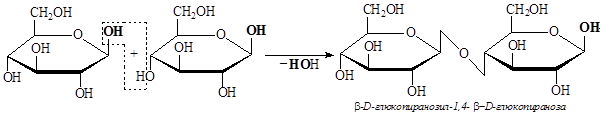
Лактоза (молочный сахар) – дисахарид, присутствующий в молоке в довольно значительных количествах: в коровьем молоке содержание лактозы составляет 4–5,5%, в женском молоке – 5,5–8,4%.
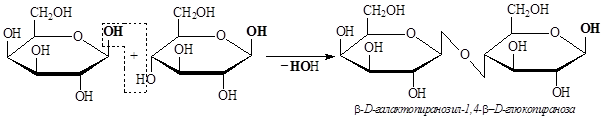
Этот дисахарид состоит из остатков b-D-галактопиранозы и b-D-глюкопиранозы, связанных b-1,4 – гликозидной связью. Лактоза отличается от всех других сахаров отсутствием гигроскопичности – она не отсыревает. Это свойство лактозы используют в фармацевтике: если нужно приготовить с сахаром какой-либо порошок, содержащий легко гидролизующееся лекарство, то берут молочный сахар; если взять другой сахар, он быстро отсыреет и легко гидролизующееся лекарственное вещество быстро разложится.
Трегалоза – дисахарид, состоящий из двух остатков a-D-глюкопиранозы, связанных между собой a-1,1-гликозидной связью.

Трегалоза найдена в пшенице, молодых грибах и водорослях. Как и фруктоза, трегалоза относится к невосстанавливающим дисахаридам.
Сахароза (тростниковый или свекловичный сахар) – дисахарид, состоящий из остатков a-D-глюкопиранозы и b-D-фруктопиранозы, связанных посредством a-1,2-гликозидной связи:
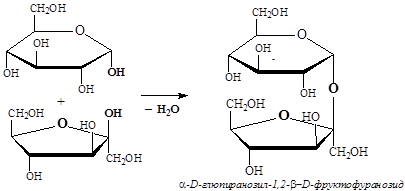
Сахароза чрезвычайно распространена в растительном мире: она содержится в листьях растений, в плодах. Кроме сахарного тростника и сахарной свеклы, большое количество сахарозы содержат клен (кленовый сок), пальма, кукуруза. Сахароза является наиболее известным и широко применяемым сахаром. Молекула сахарозы не обладает восстанавливающей активностью, так как не имеет свободной полуацетальной гидроксильной группы (они задействованы в образовании a-1,2-гликозидной связи). Растворы сахарозы не подвергаются мутаротации из-за невозможности протекания цикло-оксо-таутомерии. В реакциях сахароза проявляет свойства восьмиатомного спирта, образуя простые и сложные эфиры, а также гликозида, подвергаясь гидролизу в кислой среде с образованием глюкозы и фруктозы.
Лекция № 12
a -Аминокислоты и белки
Аминокислоты – производные карбоновых кислот, в молекулах которых один или несколько атомов водорода в углеводородном радикале замещены на одну или несколько аминогрупп. Наибольшее биологическое значение имеют a-аминокислоты, как структурные единицы белков и пептидов, b-,g- и d-аминокислоты играют гораздо меньшую роль. Большинство встречающихся в природе аминокислот имеют структуру типа R-CH(NH2)-COOH и отличаются только строением радикала R. В белках чаще всего встречается около 22 аминокислот. Для поддержания жизнедеятельности человека определенные аминокислоты должны ежедневно поступать с пищей, так как организм не синтезирует или не способен их синтезировать в достаточном количестве. Такие аминокислоты называются незаменимыми. Все аминокислоты, кроме простейшего представителя – глицина, содержат хиральный центр у a-углеродного атома. Большинство природных аминокислот принадлежит к L-ряду стереохимической номенклатуры.
Аминокислоты обычно подразделяют на три группы:
1) содержащие нейтральные неполярные (углеводородные) боковые цепи:

2) содержащие нейтральные полярные боковые цепи:
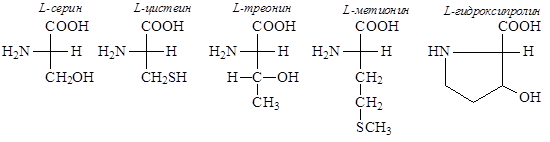

3) содержащие боковые цепи с кислотной или основной функцией:
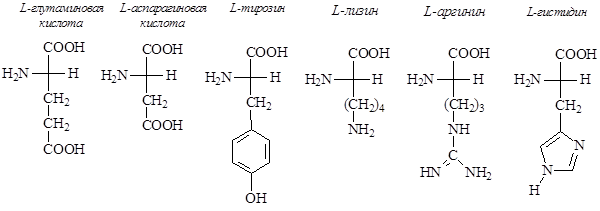
Некоторые аминокислоты (цистин, треонин, изолейцин и гидроксипролин) имеют два хиральных центра и могут существовать в виде четырех стереоизомеров. Однако для цистина число возможных стереоизомеров меньше из-за наличия плоскости симметрии у мезоформы, которая оптически неактивна.
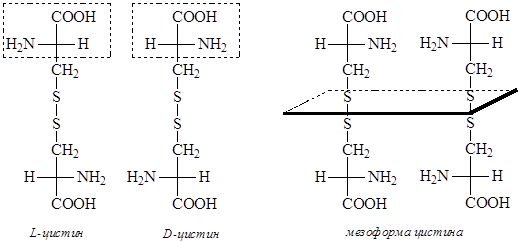
a-Аминокислоты в свободном виде представляют собой твердые кристаллические вещества, хорошо растворимые в воде, плохо растворимые в спирте, не растворимые в эфире и других неполярных органических растворителях. a-Аминокислоты характеризуются высокими температурами плавления, что обусловлено их существованием в виде цвиттер-ионов (биполярных ионов):
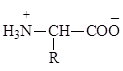
Наличие как основной (NH2), так и кислотной (СООН) групп в молекуле означает, что в водном растворе реальное строение частиц зависит от значения рН. При низких значениях рН карбоксильная группа находится в недиссоциированном состоянии, аминогруппа – в протонированной форме.
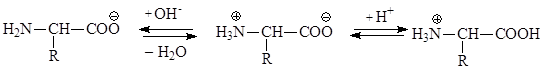
При высоких значениях рН аминокислоты в водных растворах находятся в виде аминокарбоксилат-анионов. При промежуточных значениях рН основная частица, находящаяся в растворе в существенных количествах, – это цвиттер-ион, молекулярная форма RCH(NH2)CООН не присутствует в сколько-нибудь значительной степени. На существовании в сильно кислой среде аминокислот в катионной форме и в щелочной среде – в анионной форме основано разделение аминокислот методом электрофореза: катионы под действием электрического тока перемещаются к катоду (отрицательно заряженному электроду), анионы – к аноду (положительно заряженному электроду). рН раствора аминокислоты, при котором средний заряд молекулы равен нулю, называется изоэлектрической точкой (рI). Состояние аминокислоты, в котором ее заряд равен нулю, называется изоэлектрическим состоянием (т.е. в растворе концентрация биполярных ионов аминокислоты максимальна, а концентрации катионных и анионных форм аминокислоты минимальны и равны между собой). Для моноаминомонокарбоновых кислот значения рН, при котором они находятся в изоэлектрическом состоянии, лежат в пределах 5.5-6.3, что близко к нейтральной среде, поэтому такие аминокислоты называют нейтральными. Для моноаминодикарбоновых кислот значения рН, при котором они находятся в изоэлектрическом состоянии, лежат в сильно кислой среде (рI<3.1), поэтому такие аминокислоты называют кислыми. У диаминомонокарбоновых кислот значения рН в изоэлектрическом состоянии лежат в сильно щелочной среде (рI>9.8), такие аминокислоты называют основными.
В реакциях аминокислоты проявляют себя как гетерофункциональные соединения, т.е. реагируют в зависимости от реакционного партнера либо одной функциональной группой, либо обе функциональные группы вступают в реакцию с реагентом.
Качественной реакцией на a-аминокислоты является их взаимодействие со свежеполученным гидроксидом меди (II), в результате которого образуется внутрикомплексное соединение сине-фиолетового цвета:
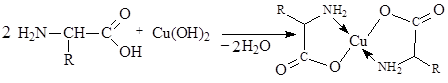
Реакции по аминогруппе:
Под действием ацилирующих агентов аминокислоты легко подвергаются ацилированию с образованием амидов:
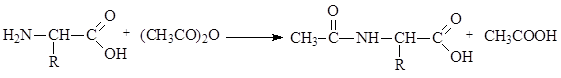 Превращение аминогруппы в амидную используется для защиты аминогруппы при синтезе пептидов и белков.
Превращение аминогруппы в амидную используется для защиты аминогруппы при синтезе пептидов и белков.
Как амины, аминокислоты подвергаются алкилированию под действием галогеналканов:
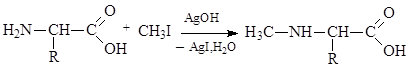
Аминокислоты реагируют с альдегидами, образуя аминоспирты, которые легко могут быть превращены в имины (основания Шиффа). Реакция аминокислот с формальдегидом приводит к образованию аминоспиртов, которые можно оттитровывать, используя метод кислотно-основного титрования. Непосредственное титрование аминокислот щелочью не может быть использовано для их количественного определения вследствие амфотерности аминокислот:
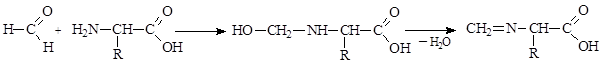 Аминокислоты могут вступать в реакции дезаминирования. В лабораторных условиях (in vitro) дезаминирование происходит под действием азотистой кислоты:
Аминокислоты могут вступать в реакции дезаминирования. В лабораторных условиях (in vitro) дезаминирование происходит под действием азотистой кислоты:
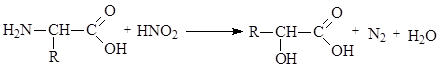
В животных и растительных организмах дезаминирование происходит с участием ферментов и может быть окислительным или неокислительным.
Неокислительное дезаминирование приводит к образованию a,b-непредельных кислот и отщеплению аммиака:
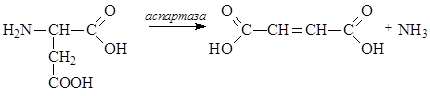
Окислительное дезаминирование идет с участием кофермента НАД+ и оксидаз – ферментов, катализирующих окислительно-восстановительные реакции.
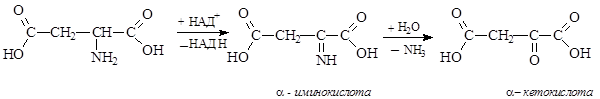
a-Аминокислоты реагируют с нингидрином с образованием продукта сине-фиолетового цвета, что используется как общая качественная реакция на a-аминкислоты:
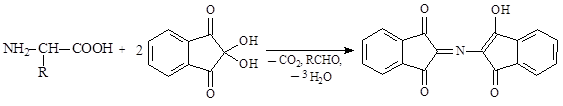
Взаимодействие a-аминокислот с 2,4-динитрофторбензолом приводит к образованию динитрофенильных производных, окрашенных в желтый цвет. Эти соединения легко идентифицируются хроматографическими методами по значениям Rf.
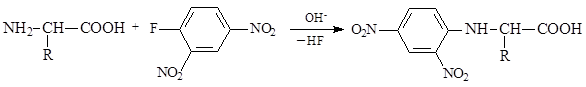
Взаимодействие с 2,4-динитрофторбензолом ранее использовалось для идентификации N-концевой аминокислоты белков и пептидов. Для этого на пептид действовали 2,4-динитрофторбензолом, а затем продукт подвергали гидролизу, получая в итоге смесь a-аминокислот и концевую аминокислоту в виде динитрофенильного производного. При помощи хроматографического анализа легко идентифицировали N-концевую кислоту по ее динитрофенильному производному и определяли аминокислотный состав пептида. Однако аминокислотная последовательность в этом случае оставалась неизвестной. Только для дипептидов, используя данный способ, можно было установить их аминокислотную последовательность.
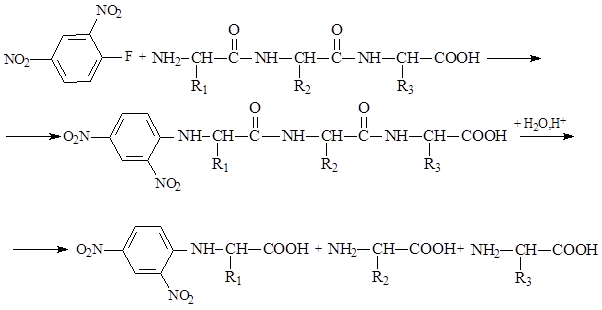
Один из универсальных способов установления аминокислотной последовательности основан на взаимодействии a-аминокислот с фенилизотиоцианатом:
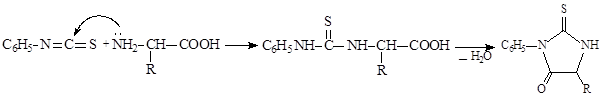
На первом этапе реакции идет нуклеофильное присоединение – аминогруппа аминокислоты присоединяется к атому углерода за счет разрыва двойной связи углерод-азот, на втором этапе происходит внутримолекулярное нуклеофильное замещение, приводящее к образованию фенилтиогидантоина – циклического амида. Для определения аминокислотной последовательности пептида его подвергают воздействию фенилизотиоцианата в щелочной среде. Как и в предыдущем примере, сначала происходит нуклеофильное присоединение к фенилизотиоцианату свободной аминогруппы N-концевой аминокислоты.
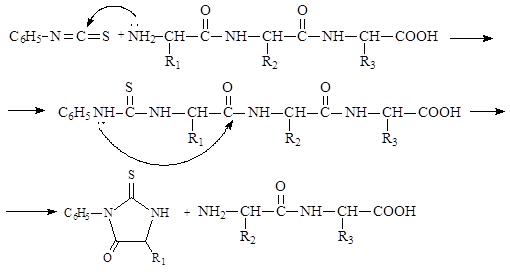
Дальнейшая обработка слабой кислотой без нагревания приводит к отщеплению от пептидной цепи N-концевой аминокислоты в виде фенилтиогидантоинового (ФТГ) производного, которое затем идентифицируется хроматографически. Последующее добавление к укороченному на одну аминокислоту пептиду фенилизотиоцианата в конечном итоге опять приводит к отщеплению аминокислоты с N-конца в виде ФТГ-производного. Таким образом можно определить аминокислотную последовательность, идентифицируя отщепляющиеся аминокислоты в виде их ФТГ-производных.
Реакции аминокислот по карбоксильной группе
Аминокислоты образуют сложные эфиры, реагируя с избытком спирта в присутствии HCl, как катализатора. Реакция протекает в спиртовой среде, а образующийся сложный эфир в виде соли выпадает в осадок. Действуя более сильными основаниями на аммониевую соль сложного эфира, можно выделить сложный эфир в свободном виде:
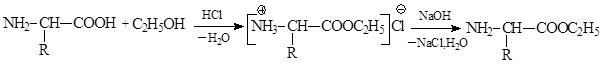 Образование сложных эфиров используется для защиты карбоксильной группы при синтезе пептидов.
Образование сложных эфиров используется для защиты карбоксильной группы при синтезе пептидов.
Карбоксильная группа аминокислот может быть превращена в ангидридную или галогенангидридную группу, но при проведении этих реакций необходимо предварительно защитить аминогруппу:
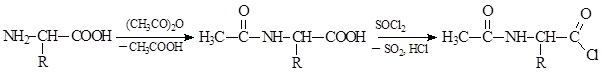
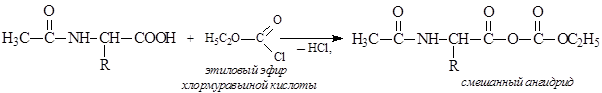
Превращение карбоксильной группы в более реакционные ангидридную или галогенангидридную группы используют для активации карбоксильной группы при синтезе белков и пептидов.
Избыток аминокислот в организме уменьшается не только за счет реакций дезаминирования, но и в результате реакций декарбоксилирования. Декарбоксилирование a-аминокислот протекает легко, что объясняется наличием у a-углеродного атома двух электроноакцепторных заместителей (NH2- и СООН-группы). Вне организма (in vitro) декарбоксилирование происходит при нагревании аминокислот в присутствии щелочей, в организме (in vivo) – с участием ферментов – декарбоксилаз, приводя к образованию биогенных аминов:
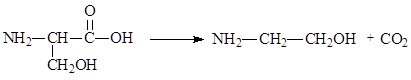

Стратегия пептидного синтеза
Пептиды и белки часто представляют как результат реакции поликонденсации a-аминокислот. В действительности же, на практике реакцию поликонденсации не используют, так как даже при синтезе дипептидов этим способом возникают проблемы с аминокислотной последовательностью и выходом нужного дипептида.
При взаимодействии двух разных аминокислот образуются семь дипептидов (без учета трипептидов, тетрапептидов и т.д.), разных по строению и аминокислотной последовательности, что существенно уменьшает количество нужного продукта.
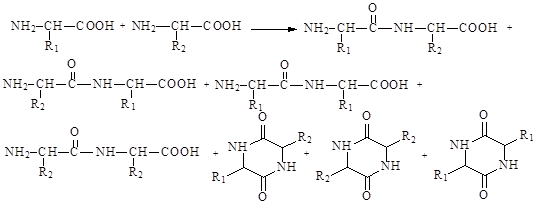
Образование пептидной связи не представляет проблемы – пептидные связи легко образуются при взаимодействии амина или аминокислоты с активированной карбоксильной группой (ангидридной или галогенангидридной) другого соединения. Проблема в синтезе белков и пептидов заключается в получении пептида или белка с нужной аминокислотной последовательностью. Чтобы преодолеть эти трудности, пептид синтезируют по стадиям, на каждой из которых приходится защищать (блокировать) одни функциональные группы и активировать другие. Активными должны быть функциональные группы, участвующие в образовании нужной амидной связи – карбоксильная группа одной молекулы с защищенной аминогруппой и аминогруппа другой молекулы с защищенной карбоксильной группой. Для защиты карбоксильной группы обычно используют ее перевод в сложный эфир, так как сложные эфиры легче гидролизуются, чем амиды, а удалить защитную группу можно, не затрагивая пептидную связь. Для защиты аминогруппы используют ее превращение в особые типы амидов, которые можно разрушать (снимать защиту) в условиях, в которых пептидные и амидные связи не затрагиваются. Для активации карбоксильной группы используют ее превращение в галогенангидридную группу или в смешанный ангидрид. Если боковые цепи аминокислот содержат реакционные функциональные группы, их также надо защищать.
Рассмотрим стратегию пептидного синтеза на примере образования аланилглицина.
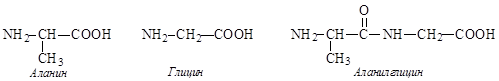
Для получения такой последовательности соединения аминокислот необходимо:
1) защитить аминогруппу аланина, превратив ее в менее реакционно- способную амидную группу действием карбобезоксихлорида:
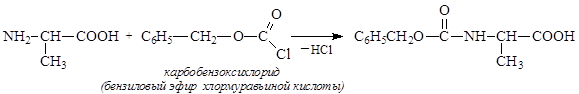
2) активировать карбоксильную группу аланина, содержащего защищенную аминогруппу, превратив ее в смешанный ангидрид действием этилхлорформиата:
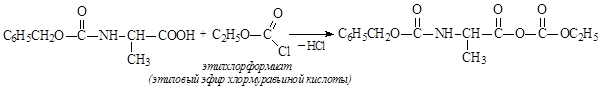 3) защитить карбоксильную группу глицина, переведя ее в сложноэфирную:
3) защитить карбоксильную группу глицина, переведя ее в сложноэфирную:
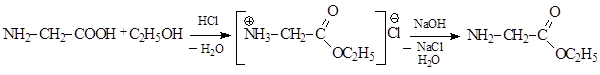 4) провести реакциию образования пептида:
4) провести реакциию образования пептида:
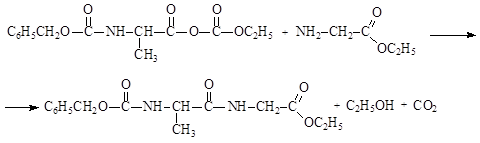
5) удалить защитные группы в полученном дипептиде; карбобензокси-группа удаляется гидрированием, сложноэфирная группировка удаляется действием растворов щелочей:
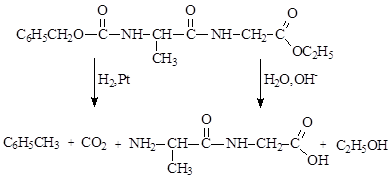
Основной интерес химиков и биологов сосредоточен на установлении взаимосвязи строения и функции белка. Пептиды и белки могут содержать в молекуле как основные (-NH2, -СОО-), так и кислотные (-NH3+, -СООН) функциональные группы, расположенные на конце полиамидной цепи, либо включенные в полифункциональные аминокислоты. Общий заряд на молекуле меняется в зависимости от рН среды точно так же, как и в случае аминокислот. Так, при низких значениях рН молекула белка имеет положительный заряд, в то время как при высоких значениях рН на молекуле возникает отрицательный заряд. Неудивительно, что свойства белковых растворов заметно меняются при переходе через изоэлектрическую точку, поскольку сольватация или взаимодействие с соседними белковыми молекулами зависят, вероятно, и от распределения заряда на поверхности молекулы, и от ее суммарного заряда. Вязкость раствора желатина проходит через минимум при рН = 4,7 (изоэлектрическая точка), растворимость инсулина и казеина – наименьшая в их изоэлектрических точках (5,3 и 4,7, соответственно). Зависимость суммарного электрического заряда на полипептиде или белке от значения рН среды используется для разделения белков с помощью электрофореза.
Известно, что активность многих ферментов зависит от рН среды и достигает максимума при его определенном значении. Считается, что ферменты адсорбируют субстраты на особой части молекулы, называемой «активный центр». Изменение рН приводит к перераспределению зарядов на молекуле, что, в свою очередь, меняет ее гидратацию либо за счет изменения числа групп, связанных водородными связями, либо из-за разной степени ассоциации молекул воды вокруг белковой молекулы, осуществляемой в результате взаимодействия с заряженными участками. Кроме того, сами рецепторные группы активного центра фермента, присоединяющие субстрат, в зависимости от рН могут находиться в протонированном или депротонированном состояниях. Все перечисленные эффекты могут снижать легкость адсорбции ферментом своего особого субстрата, тем самым уменьшая его каталитическую активность.
На биологическую активность белков влияет не только среда; их функции существенным образом зависят от их строения. Обычно структурные особенности белков разделяют на несколько категорий. Первичная структура белка – это последовательность аминокислотных остатков в цепи, которая устанавливается с помощью химических методов анализа. Цепь может сворачиваться в спираль или принимать особую форму за счет образования водородных связей между амидными группами. Эта особенность структуры белка, являющаяся результатом взаимодействия между амидными (пептидными) связями, называется вторичной структурой. Дальнейшее свертывание вторичной структуры является результатом взаимодействия функциональных групп боковых цепей аминокислот (-SH, -NH2, -ОН, -СООН и т.д.), что и составляет третичную структуру. Таким образом, вторичная и третичная структура представляют собой упорядоченное расположение полипептидной цепи в пространстве. Однако вторичная структура закрепляется за счет водородных связей между пептидными группами, а третичная – за счет электростатических и гидрофобных взаимодействий. Белки могут обладать четвертичной структурой, обусловленной взаимодействием нескольких белковых молекул, приводящим к образованию групп или пучков молекул, которые могут обладать высокой степенью симметрии. Эти образования иногда можно обнаружить с помощью электронного микроскопа.
Многие полипетиды и белки исследовались с помощью рентгеноструктурного анализа. При этом были подтверждены некоторые характерные особенности их структуры. Наиболее часто встречаются два типа организованной вторичной структуры, хотя нередко молекулы белков имеют беспорядочное строение. В a-форме полиамидная цепь свернута в спираль, в которой расположенные рядом витки соединяются за счет водородных связей между соседними амидными группами. Спираль имеет правое вращение, при этом объемные боковые цепи L-аминокислот направлены от центра спирали.
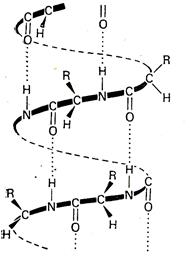
В b-структуре полиамидные цепи располагаются рядом в антипараллельном положении, образуя слой полипептидных цепей, связанных между собой водородными связями. При таком строении боковые цепи полипептидных молекул лежат попеременно над и под плоскостью слоя.
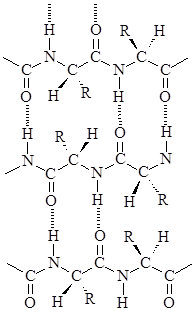
Участки b-структуры могут образоваться и внутри одной белковой молекулы за счет складывания свернутой полипептидной цепи. Белки, имеющие развернутую b-структуру (например, фиброин шелка), растягиваются с трудом, поскольку полипептидные цепи уже полностью вытянуты, тогда как белки с преобладающей a-структурой (например, волосы, шерсть) – эластичны, так как механическое напряжение в этом случае снимается за счет превращения спиралевидной полипептидной цепи в растянутую конформацию b-структуры.
Третичная структура белков, обусловленная взаимодействием боковых цепей аминокислот, не приводит к такой высокой упорядоченности структуры, как в предыдущем случае. Помимо водородных связей, важным фактором стабилизации третичной структуры является образование дисульфидных связей. Третичная структура часто придает белковой молекуле такую конформацию, при которой гидрофильные группы расположены на поверхности молекулы, а гидрофобные группы направлены внутрь, к центру молекулы.
Четвертичная структура белков варьируется очень широко. Например, общей формой четвертичной структуры обладают волокнистые белки (волосы, шерсть). Эта структура состоит из шести белковых цепей, каждая из которых имеет форму a-спирали, закрученной вокруг центральной спиралеобразной белковой молекулы. В результате образуется веревкообразная структура.
Биологическая активность белков нередко тесно связана с высокой организацией структуры, и живые организмы синтезируют белки требуемой конформации, которая часто оказывается метастабильной (не самой устойчивой из всех возможных структур). Под влиянием нагревания, изменения значений рН, химических реагентов, ультрафиолетового света и других воздействий белки часто теряют свою биологически необходимую конформацию, превращаясь в случайные неорганизованные структурные единицы и утрачивая свою биологическую активность. Такой процесс называется денатурацией. Денатурация оказывает особо глубокий эффект на активность ферментов. По этой причине моющие порошки, содержащие ферменты, лучше всего применять при низкой температуре, так как при высокой температуре прежде всего проявляется действие входящих в их состав детергентов.
ЛЕКЦИЯ № 13
Нуклеиновые кислоты
Нуклеиновые кислоты (ДНК и РНК) представляют собой макромолекулы кислотного характера, содержащиеся в основном в ядре клетки, но также встречающиеся в цитоплазме. Углеводы и белки представляют собой материал для построения живых систем и их жизнедеятельности, в то время как нуклеиновые кислоты служат источником генетической информации, направляющей все эти процессы. Соединясь с белками, нуклеиновые кислоты образуют нуклеопротеины. Установлено, что вирусы, которые в некоторых случаях можно выделить в кристаллическом виде, являются большими нуклеопротеинами.
Гидролиз нуклеиновых кислот дает три типа продуктов: фосфорную кислоту, углевод (рибоза или дезоксирибоза) и группу азотистых оснований (урацил, тимин, цитозин, аденин, гуанин). ДНК и РНК отличаются по строению моносахарида, входящего в состав нуклеиновых кислот – в состав ДНК входит дезоксирибоза, в состав РНК – рибоза:
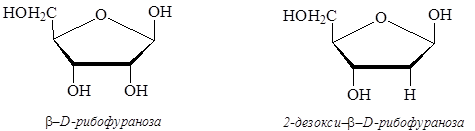
Существуют также некоторые различия в азотистых основаниях, входящих в состав нуклеиновых кислот: аденин, гуанин, цитозин образуются при гидролизе и ДНК, и РНК, в качестве четвертого основания ДНК содержат тимин, РНК – урацил. Урацил, тимин и цитозин представляют собой производные пиримидина:
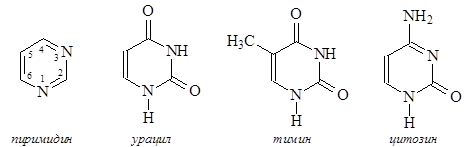
Аденин и гуанин являются производными пурина:
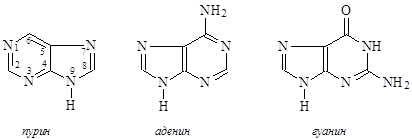
Для кислородсодержащих азотистых оснований характерна лактам-лактимная таутомерия, а для пуриновых оснований – прототропная:
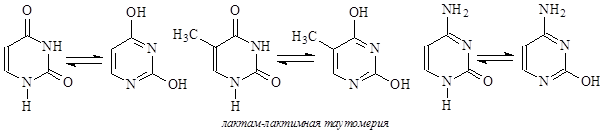
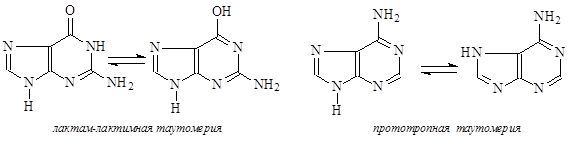
В нуклеиновых кислотах каждое азотистое основание связано через атом азота N-гликозидной связью с аномерным атомом углерода рибозы или дезоксирибозы. При этом нужно отметить, что всегда образуется b-гликозидная связь. Этот блок – углевод-азотистое основание – называется нуклеозидом.
При названии нуклеозидов у азотистых оснований пиримидинового ряда окончания заменяют на -идин, у оснований пуринового ряда – на окончание -озин.
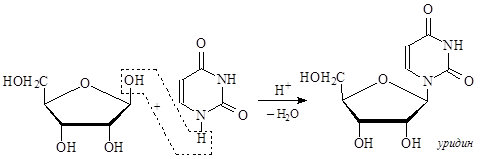
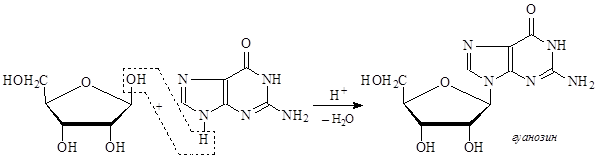
Кроме того, нуклеозиды, образованные с участием дезоксирибозы, получают приставку дезокси.
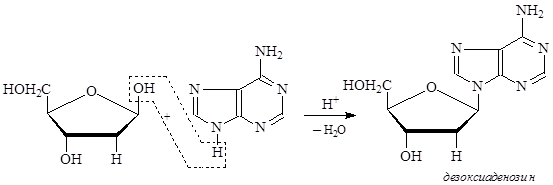
Так как тимин входит в состав только ДНК, в виде исключения нуклеозид, образуемый им и дезоксирибозой, называют тимидин, а не дезокситимидин.
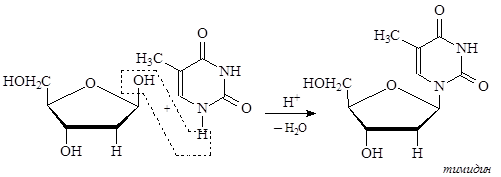
Нуклеозиды являются N-гликозидами, и как гликозиды (ацетали), подвергаются гидролизу в кислой среде, но устойчивы к гидролизу в слабощелочных средах ( в сильно щелочной среде происходит не гидролиз, а разрушение гетероциклов).
Нуклеозиды за счет гидроксильных групп рибозы или дезоксирибозы могут реагировать с кислородсодержащими кислотами с образованием сложных эфиров. Эфиры нуклеозидов и фосфорной кислоты называются нуклеотидами. Сложноэфирная связь может образовываться между остатком фосфорной кислоты и гидроксильной группой в положении 5¢- или 3¢-рибозы или дезоксирибозы. Названия нуклеотидам дают либо как сложным эфирам фосфорной кислоты, либо как кислотам, так как остаток фосфорной кислоты содержит гидроксильные группы, за счет которых нуклеотиды могут выступать в качестве кислот.
В названии нуклеотидов, как эфиров, сначала называется нуклеозид, затем указывается положение остатка фосфорной кислоты (3¢ или 5¢) и добавляется название кислотного остатка – фосфат. В названиях нуклеотидов как кислот сначала указывают положение сложноэфирной связи (3¢ или 5¢), затем идет название азотистого основания с добавлением окончания «овая кислота»:
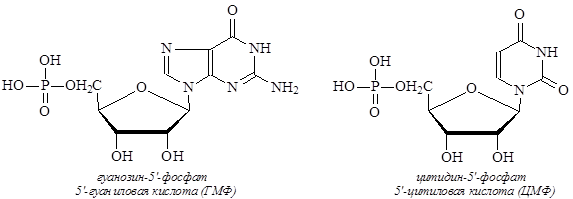

Существуют нуклеотиды, в которых две гидроксильные группы (в положениях 3¢ и 5¢) этерифицированы фосфорной кислотой. Такие нуклеотиды называют циклофосфатами. Почти во всех клетках имеются два циклофосфата – цАМФ и цГМФ.
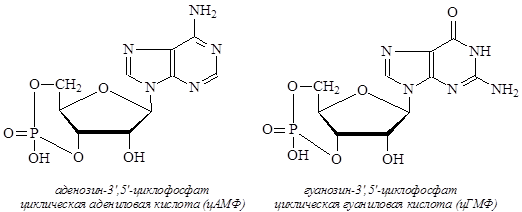
Циклофосфаты участвуют в регуляции внутриклеточных процессов, способны превращать ряд инертных белков в ферменты.
Как сложные эфиры, нуклеотиды подвергаются гидролизу как под действием кислот, так и под действием щелочей. При щелочном гидролизе образуются соль фосфорной кислоты и нуклеозид (гликозидная связь не подвергается щелочному гидролизу), при кислотном гидролизе получаются фосфорная кислота, углевод и азотистое основание.
ДНК и РНК построены по общему принципу: мононуклеотиды соединены в нуклеиновых кислотах за счет остатков фосфорной кислоты, связывающей 5-й атом углерода рибозы или дезоксирибозы одного мононуклеотида с 3-м атомом углерода другого мононуклеотида, т.е нуклеиновые кислоты представляют собой полиэфиры N-гликозидов – нуклеозидов.
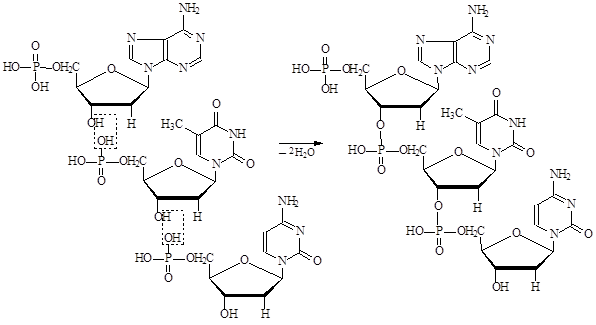
Как видно из примера образования тринуклеотида, полимерная цепь нуклеиновых кислот состоит из чередующихся углеводных и пентозных остатков, а азотистые основания являются «боковыми группами», присоединенными к остатку моносахарида. В нуклеиновых кислотах выделяют 5¢-конец, на котором находится остаток фосфорной кислоты, образующий только одну сложноэфирную связь с гидроксильной группой в положении 5¢-моносахарида (этот конец цепи обозначают в сокращенной записи буквой «ф»), и 3¢-конец, на котором находится свободная гидроксильная группа в положении 3¢-моносахарида. Последовательность нуклеотидных звеньев в приведенном примере можно записать в сокращенном виде следующим образом: фдАфдТфдЦ. Буква ф ставится справа от нуклеотида, если он этерифицирован по гидроксильной группе третьего атома углерода моносахарида, если нуклеотид образует сложноэфирную связь за счет пятого атома углерода в моносахариде, то буква ф ставится слева. Последовательность нуклеотидных звеньев в цепи ДНК или РНК называется первичной структурой.
Пространственная организация полинуклеотидной цепи называется вторичной структурой. Вторичная структура ДНК представляет собой двойную спираль, состоящую из двух переплетенных цепей ДНК. В полный виток каждой спирали укладывается около десяти нуклеотидных единиц и две цепи выстраиваются в противоположных направлениях так, чтобы азотистые основания были направлены внутрь двойной спирали. Причем, остаток тимина в одной цепи образует всегда водородные связи с остатком аденина другой цепи, а остаток цитозина – с остатком гуанина. Это подтверждается данными гидролиза, которые показывают, что в ДНК соотношение тимин:аденин и цитозин:гуанин равно 1:1. Эти основания называют комплементарными. Две цепи ДНК, образующие двойную спираль, не идентичны, но комплементарны между собой, т.е. нуклеотидная последовательность одной цепи определяет первичную структуру второй цепи.
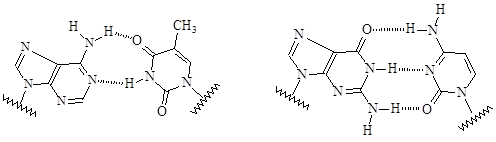
Между остатками аденина и тимина образуются две водородные связи, между остатками гуанина и цитозина – три. Водородные связи между комплементарными основаниями являются одним из видов взаимодействий, стабилизирующих двойную спираль.
Клетки живого организма постоянно делятся. Когда происходит деление клетки, необходимо точное воспроизведение молекулы ДНК. Этот процесс называется репликацией и происходит следующим образом. Две цепи двойной спирали начинают разделяться и на каждой из первоначальных цепей идет биосинтез новой цепи с учетом комплементарности азотистых оснований. Вновь образованные цепи не идентичны, но комплементарны исходным цепям, служившим им матрицей. В итоге получаются две новые двойные спирали, состоящие из одной синтезированной и одной матричной цепей. Подобным образом на деспирализованной цепи ДНК идет образование матричной РНК, которая затем сама является матрицей для биосинтеза белка. Генетическая информация записана в нуклеотидной последовательности в ДНК. Изменение нуклеотидной последовательности в ДНК приведет к изменению нуклеотидной последовательности в м-РНК, а это может привести к изменению аминокислотного состава синтезируемого белка. Такое изменение ДНК называется мутацией. Накопление мутаций приводит к возрастанию числа ошибок в биосинтезе белка. Последствия мутаций иногда помогает избежать вырожденность генетического кода, в результате чего многие мутации, например изменение последнего основания в кодоне, не изменяют аминокислотного состава белка, т.е происходит кодирование тех же аминокислот или аминокислот с похожей боковой цепью. Однако изменение даже одного основания в ДНК может иметь и серьезные последствия. Так, например, у больных серповидноклеточной анемией гемоглобин менее растворим, чем нормальный, и осаждается в тонких кровеносных сосудах, вызывая сильную боль. Такой аномальный гемоглобин разрушается селезенкой, что приводит к анемии. Оказалось, что серповидноклеточная анемия вызвана изменением одного азотистого основания в ДНК, в результате чего м-РНК вместо глутаминовой кислоты кодирует валин, который далее включается в состав гемоглобина, что и приводит к образованию аномального гемоглобина с особыми свойствами.
Мутации также могут быть обусловлены случайными ошибками при копировании ДНК при подготовке клетки к делению; кроме того, они могут быть вызваны действием химических препаратов и различными излучениями (ионизирующими и неионизирующими, корпускулярными и некорпускулярными).
Большое количество соединений обладает мутагенными свойствами, среди них широко известны азотистая кислота, гидроксиламин, различные алкилирующие агенты – N-нитрозо-N-этилмочевина, серные, азотистые иприты и др.
Действие азотистой кислоты приводит к дезаминированию азотистых оснований:
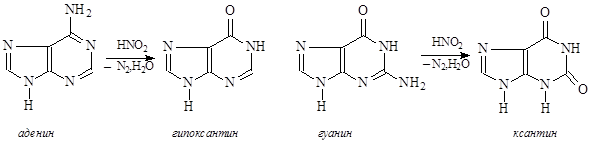
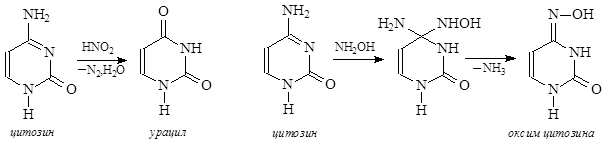
Аденин превращается в гипоксантин, гуанин – в ксантин, цитозин – в урацил (на тимин HNO2 не действует). Это приводит к замене в ДНК комплементарных пар оснований, так как аденин комплементарен тимину, а гипоксантин образует комплементарную пару с цитозином, цитозин комплементарен гуанину, а урацил образует комплементарную пару с аденином.
Действие гидроксиламина сводится главным образом к реакции с цитозином – в нейтральных и слабокислых средах образуется производное, которое комплементируется с аденином.
Многие алкилирующие соединения обладают выраженным мутагенным действием. Для соединений этого типа характерна реакция алкилирования по седьмому атому азота гуанина, в результате протекания которой может происходить отщепление азотистого основания от остатка дезоксирибозы.
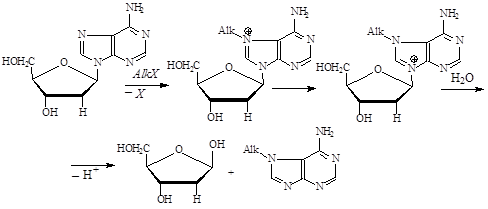
Велико также значение реакций алкилирования оснований, не сопровождающихся выщеплением их из ДНК. Такие процессы происходят при алкилировании аденина по первому и третьему атомам азота, тимина – по первому атому азота и т.д. Эти процессы существенно искажают комплементирование оснований при синтезе ДНК. Для ипритов, помимо описанных выше реакций, характерно также образование мостиков между азотистыми основаниями, сшивающих либо разные цепи ДНК, либо основания одной цепи. Возможно образование сшивок ДНК-белок. Именно эта группа алкилирующих агентов часто вызывает хромосомные перестройки.
Излучения являются мутагенным фактором со сложным механизмом действия. В зависимости от природы излучения ведущие механизмы могут быть различными, однако ни в одном случае не удается объяснить наблюдаемые эффекты каким-то одним типом реакций.
Для ионизирующих излучений характерно не только прямое воздействие на ДНК (первичное), но и непрямое, состоящее в ионизации других молекул, особенно воды, с образованием свободных радикалов, которые далее воздействуют на ДНК. Свободные радикалы могут вызывать, во-первых, различные окислительные процессы, в число которых входит дезаминирование, гидроксилирование цитозина, тимина, аденина, образование гидроперекисей тимина и, наконец, окисление остатка дезоксирибозы. Во-вторых, реакции окисления сочетаются с разрывами пиримидиновых и в меньшей степени пуриновых циклов, разрывами связей между дезоксирибозой и остатком фосфорной кислоты, дезоксирибозой и основаниями, разрывом связей углерод-углерод в самой дезоксирибозе.
Ультрафиолетовое излучение, относящееся к некорпускулярным излучениям, хотя обладает низкой проникающей способностью, также является мутагенным фактором. Кванты УФ-излучения поглощаются нуклеиновыми кислотами, что приводит к переходу молекул в возбужденное состояние, повышению их реакционной способности и протеканию реакций, приводящих к повреждению оснований. Особое значение приобретает эффект образования димеров тимина:
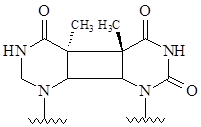
В неденатурированных участках молекулы ДНК димеризация происходит лишь между соседними остатками тимина одной цепи. Тиминовые димеры располагаются вдоль цепи ДНК не случайным образом, а группируются так, что на небольшом участке цепи ДНК сосредотачивается в среднем по 6 димеров. Этого достаточно, чтобы создать препятствия при репликации и спровоцировать либо выпадение целых участков, либо искажение транскрипции.
Рекомендуемая литература
Основная литература
1. Овчинников Ю.А. «Биоорганическая химия».- М., 1987 г.
2. Романовский И.В. «Основы биоорганической химии».- Мн.,1999 г.
3. «Руководство к лабораторным занятиям по биоорганической химии». – Под редакцией Н.А. Тюкавкиной.- М., 1999 г.
4. «Руководство к лабораторным занятиям по биоорганической химии». Под редакцией Н.А. Тюкавкиной.- М., 1985 г.
5. «Руководство к практикуму по биоорганической химии». Под редакцией И.В.Романовского. – Мн., 2003 г.
6. Тюкавкина Н.А., Бауков Ю.И. «Биоорганическая химии».- М., 1991 г.
Дополнительная литература
7. Николаев А.Я. «Биологическая химия». – М., 1989 г.
8. Потапов В.М. «Стереохимия». – М., 1988 г.
9. Тейлор Г. «Основы органической химии».- М., «Мир». – 1989 г.
10. Терней А. «Современная органическая химия». – М., 1981 г.
Содержание
| Лекция №1. Предмет и задачи биоорганической химии. Взаимное влияние атомов и способы его передачи в органических молекулах. Сопряженные системы, ароматичность, электронные эффекты заместителей | |
| Лекция №2. Основы реакционной способности органических соединений.Классификация органических реакций по разным признакам. Гомолитические и гетеролитические реакции | |
| Лекция №3. Реакции электрофильного замещения в ряду ароматических соединений | |
| Лекция №4. Кислотно-основные свойства органических соединений, ионизация | |
| Лекция №5. Конкурентные реакции нуклеофильного замещения и элиминирования у насыщенного атома углерода | |
| Лекция №6. Реакции нуклеофильного присоединения (АN) к карбонильным соединениям. Окисление и восстановление органических соединений | |
| Лекция №7. Реакции нуклеофильного замещения в ряду карбоновых кислоти их производных | |
| Лекция №8. Липиды, классификация, отдельные представители. Фосфолипиды как структурные компоненты клеточных мембран. Пероксидное окисление липидов | |
| Лекция №9. Поли- и гетерофункциональные соединения. Биологически активные гетероциклические соединения. Таутомерия | |
| Лекция №10. Стереохимия органических соединений. Энантиомерия и диастереомерия | |
| Лекция №11. Углеводы. Моно- и олигосахариды | |
| Лекция №12. a -Аминокислоты и белки | |
| Лекция №13. Нуклеиновые кислоты | |
| Литература |
Учебное издание
Павловский Николай Дмитриевич
ЛЕКЦИИ ПО БИООРГАНИЧЕСКОЙ ХИМИИ
Пособие
Ответственный за выпуск В.В.Воробьев
Компьютерная верстка А.В.Яроцкая
Корректор Л.С.Засельская
Подписано в печать 08.12.2011. Формат 60х84/16. Бумага офсетная.
Гарнитура Arial Narrow. Ризография.
Усл. печ. л. 11,6. Уч.-изд. л. 7,1. Тираж 99экз. Заказ 231.
Издатель и полиграфическое исполнение
учреждение образования
«Гродненский государственный медицинский университет».
ЛИ № 02330/0548511 от 16.06.2009. Ул. Горького, 80, 230009, Гродно.
Дата добавления: 2015-08-08; просмотров: 6579;