ГЛАВА XI. Наследственные нарушения умственного и физического развития
11.1 Роль генетических факторов в возникновении расстройств речи
Возникновение большинства расстройств речи гипотетически связано с влиянием наследственных факторов. На этот факт указывают семейный характер этих патологических состояний и конкордантность по ним у монозиготных близнецов. В некоторых случаях известна генетическая природа речевых нарушений.
Алалия – отсутствие или недоразвитие речи у детей при нормальном слухе и первично сохраненном интеллекте. Одна из наследственных форм этого патологического состояния – вербальная диспраксия – имеет аутосомно-доминантный тип наследования и обусловлена мутациями гена FOXP2 (HSA7q31), который кодирует транскрипционный фактор семейства Forkhead.
Ринолалия (гнусавость) – изменение тембра голоса и искаженное произношение звуков, вызванное нарушением резонаторной функции носовой полости. Одной из причин этого дефекта звукопроизношения является врожденное расщепление неба (Рисунок XI, 1) – симптом, который присутствует при некоторых хромосомных аберрациях (разделы 5.8 и 5.9). В некоторых популяциях сочетания мутантных аллелей (гаплотипы) гена GAD1 (HSA2q31), который кодирует глутаматкарбоксилазу 1, приводят к расщеплению неба.

Рисунок XI, 1. Расщепление неба. По материалам сайта http://ru.wikipedia.org/wiki/Файл13900470_3PREOPERATION0.jpg
Предрасположенность к дислексии – нарушению способности к обучению читать и писать при нормальном интеллекте - аутосомно-доминантный признак, обусловленный мутациями в гене DYX1C1 (HSA15q21), продукт которого отвечает за взаимодействие рецептора эстрогена с белками теплового шока Hsp70 и Hsp90. На проявление этого признака также оказывают влияние гены, расположенные на HSA6 и HSA11.
Миссенс-мутация в гене N-ацетилглюкозамин-1-фосфаттрансферазы GNPTAB (HSA12q23.3) является причиной заикания. Кроме того, существует еще одна наследственная форма этого речевого нарушения, предполагаемый локус которого – HSA18p11.3-p11.2.
11.2 Наследственные формы интеллектуальных нарушений
Умственная отсталость (интеллектуальная недостаточность), выражающаяся в стойком нарушении познавательной деятельности, характерна для многих хромосомных и генных болезней как симптом.
Наиболее типичные хромосомные аномалии, при которых наблюдается умственная отсталость - синдром Дауна, синдром Шерешевского-Тернера, синдром Клайнфельтера и сидром Мартина-Белл - описаны в разделах 5.8 и 5.9.
Умственная отсталость характерна для фенилкетонурии, некоторых мукополисахаридозов и болезни Нимана-Пика (раздел9.3).
Гомоцистинурия – накопление метионина и гомоцистина по причине недостаточности фермента печени цистатионинсинтетазы, которое приводит к поражению костной ткани и ЦНС. У больных детей отмечается задержка роста, умственная отсталость, судороги, остеопороз, эктопия хрусталика, склонность к тромбозам. Общий вид больных напоминает синдром Марфана (раздел 9.3), главной отличительной особенностью является умственная отсталость. Заболевание прогрессирует быстро, больные обычно умирают в молодом возрасте. Тип наследования этого заболевания – аутосомно-рецессивный. Генетическая природа гомоцистинурии – различные мутации гена CDS (HSA21q22.3), который кодирует цистатионинсинтетазу.
Истинная микроцефалия – аутосомно-рецессивное заболевание, проявляющееся в меньшем объеме головного мозга и меньшем размере мозгового черепа, чем у здорового индивидуума (Рисунок XI, 2). При истинной микроцефалии в отличие от синдромов, при которых она присутствует в качестве симптома, отсутствуют пороки скелета и неврологические нарушения (кроме умственной отсталости). Причиной заболевания являются мутации гена ASPM (HSA1q31), кодирующего белок аномального веретена деления, который участвует в пролиферации эмбриональных фибробластов. Интересно, что впервые этот ген был описан у плодовой мушки Drosophila melanogaster (глава II), причем функция его та же, что и у человека. Это пример использования эволюционного консерватизма (раздел 8.3) в медицинской генетике.

Рисунок XI, 2. Истинная микроцефалия. По материалам http://ru.wikipedia.org/wiki/Файл:IMGP2147.JPG.
Обтурационная гидроцефалия (гидроцефалия, вызванная наследственным стенозом Сильвиева водопровода) – X-сцепленное рецессивное заболевание с прогредиентной неврологической симптоматикой, вызванное нарушением оттока цереброспинальной жидкости из первых трех мозговых желудочков. Молекулярно-генетическая основа заболевания – мутация в экзоне 22 гена L1CAM (HSAXq28), который кодирует один из белковых доменов молекулы клеточной адгезии L1.
Синдром Сотоса (мозговой гигантизм) – аутосомно-доминантный гигантизм,не связанный с гормоном роста. До 4-5 лет ребенок с таким синдромом растет почти вдвое быстрее, чем обычный. Характерны задержка развития и не прогрессирующая умственная отсталость. У больных большие кисти и стопы с утолщенным слоем подкожной жировой ткани. Голова большая, нижняя челюсть выступает вперед, глаза широко расставлены (Рисунок XI, 3). Причиной заболевания могут быть микроделеции (раздел X, 1) в районе HSA5q35 либо мутации расположенного в том же хромосомном районе гена NSD1, который кодирует ассоциированный с андрогеновым рецептором корегулятор 267.
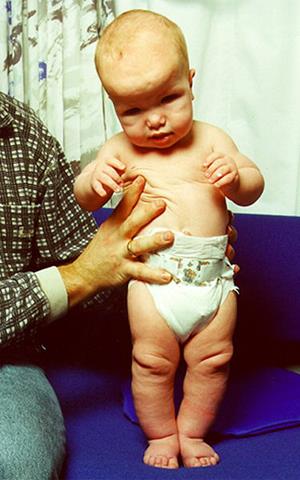
Рисунок XI, 3.Синдром Сотоса. По материалам сайта http://en.wikipedia.org/wiki/File:Sotos.jpg
Синдром Смита-Магениса характеризуется умственной отсталостью, гиперактивностью поведения, резко повышенной сонливостью, черепно-лицевыми аномалиями, наличием широких коротких рук, склонностью к нанесению повреждений самому себе. Синдром обусловлен микроделецией величиной 3,7 млн.п.н. в районе HSA17p11.2 или мутациями локализованного в том же районе индуцируемого ретиноидной кислотой гена 1 - RAI1.
Синдром Вильсона (синдром «лица эльфа») – аутосомно-доминантное заболевание, вызванное делециями величиной 1,5 – 2,5 млн.п.н. в районе HSA7q11.23. Больные имеют особое строение лица, называемое «лицом эльфа», поскольку оно напоминает этих мифологических персонажей в их классическом понимании. Для этого синдрома характерны широкий лоб, разлёт бровей по средней линии, опущенные вниз полные щёки, большой рот с полными губами (особенно нижней), плоская переносица, нос с плоским тупым концом, маленький, слегка заострённый подбородок (Рисунок XII, 4). Окраска глаз обычно ярко-голубая, со звёздчатой картиной радужки и склерами синеватого цвета. Для этого синдрома характерен дефицит наглядно-образного мышления и слабые вербальные способности.

Рисунок XI, 4.Синдром Вильсона. По материалам сайта http://beehive.thisishull.co.uk/default.asp?WCI=SiteHome&ID=8457
Туберозный склероз (болезнь Бурневилля) – аутосомно-доминантное полисистемное заболевание, при котором во множестве органов и тканей образуются доброкачественные опухоли. Повреждения головного мозга, которые происходят обычно на границе серого и белого вещества, могут вызвать эпилепсию, снижение интеллекта. Характерные новообразования кожи лица и глазного дна могут быть использованы при начальной диагностике (Рисунок XI, 5). Мутации в генах TSC1 (HSA9q34) и TSC2 (HSA16q13.3), которые кодируют соответственно гамартин и туберин, являются причиной этого заболевания.
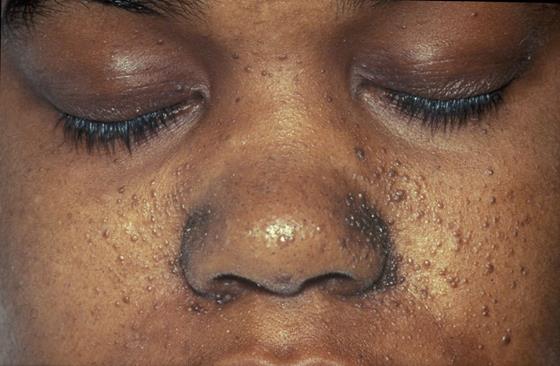
Рисунок XI, 5.Повреждения кожи при туберозном склерозе. По материалам сайта http://en.wikipedia.org/wiki/File:TuberSclerosisCase-143.jpg
Синдромы Прадера-Вилли и Ангельмана обусловлены микроделецией в районе HSA15q11-q13. Если аберрантная хромосома приходит от отца, развивается синдром Прадера-Вилли (ожирение, склонность к перееданию, гипотонус, нарушение координации движений, маленькие кисти и стопы, низкий рост, повышенная сонливость, косоглазие; пониженная плотность костей, гипогонадизм, речевая задержка, задержка психического развития, отставание в освоении навыков общей и мелкой моторики) (Рисунок XI, 6). Если от матери – синдром Ангельмана (размер головы меньше среднего, нередко с уплощением затылка, задержка в развитии навыков общей моторики, задержка речевого развития, дефицит внимания и гиперактивность, сложности с обучением, часто эпилепсия, необычные движения - мелкий тремор, хаотические движения конечностей, частый смех без повода, ходьба на негнущихся ногах) (Рисунок XI, 7). Различия в метилировании цитозина в мужском и женском организмах приводят к различному проявлению одной и той же мутации в зависимости от того, кто из родителей передал аномальную хромосому ребенку. Такое явление называется геномный импринтинг.
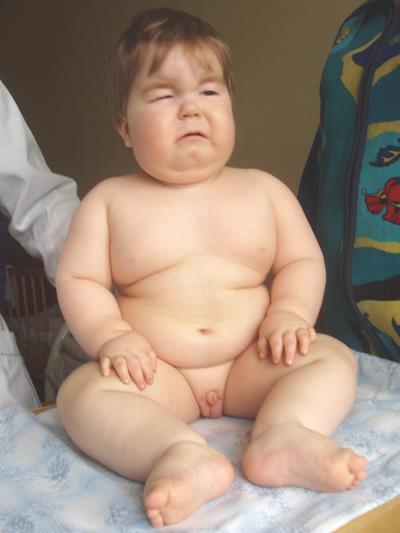
Рисунок XI, 6.Синдром Прадера-Вилли. По материалам сайтаhttp://www.medico.ru/cgi-bin/medatlas/comment.pl?rlink=&publ=1115220228_01_168&page=
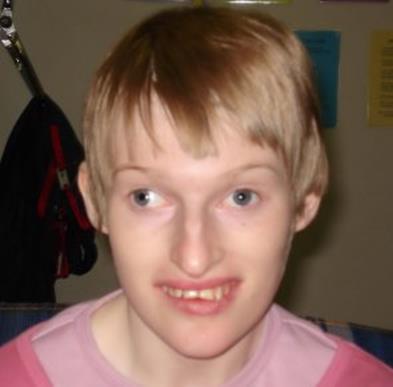
Рисунок XI, 7.Синдром Ангельмана. По материалам сайта http://www.primehealthchannel.com/angelman-syndrome-symptoms-pictures-causes-life-expectancy-and-treatment.html
Синдром Корнелии де Ланге – аутосомно-доминантное заболевание, при котором больные отстают в росте и массе тела, имеют своеобразное строение лица (густые сросшиеся брови, длинные густые ресницы, короткий нос с развернутыми ноздрями и сдавленным переносьем) и мозгового отдела черепа (микроцефалия, брахицефалия) (Рисунок XI, 8). Для больных характерны небольшие кисти, синдактилия стоп, гипертрихоз туловища и конечностей, мраморная кожа, мышечная гипотония. Пациенты часто страдают заболеваниями верхних дыхательных путей, почти у всех наблюдается умственная отсталость. Причина заболевания – мутации в гене BIPBL (HSA 5p13.1), который кодирует делангин.
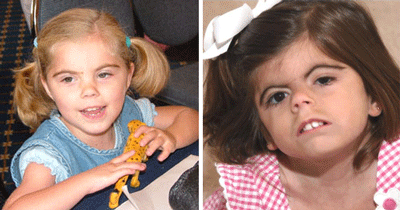
Рисунок XI, 8.Синдром Корнелии де Ланге. По материалам сайта http://kidneeds.ru/de-Lange/sindrom-kornelii-de-lange.html.
Синдром Рубинштейна-Тейби (амстердамская карликовость)- аутосомно-рецессивное заболевание, при котором длина и масса тела больных значительно отстают от нормы, череп уменьшен, брахицефальной структуры, наблюдаются аномалии строения верхних конечностей: кисти небольших размеров, короткий II палец и проксимально расположенный I палец, искривленный V. Нередко отмечается синдактилия стоп (Рисунок XI, 9). На коже у больных, кроме гипертрихоза, резко выраженного в области спины и поясницы, нередко отмечается общая мраморность, характерны краснота кончика носа, цианоз носогубной области. Умственная отсталость определяется практически у всех больных с данным синдромом. Иногда наблюдается стремление к аутоагрессии и склонность к стереотипным движениям. Для этого синдрома характерна генетическая гетерогенность – причиной могут быть мутации в гене CREBP (HSA16p13.3) и в гене EP300 (HSA22q13), которые кодируют CREB-связывающий белок и гистон-ацетилтрансферазу, соответственно.
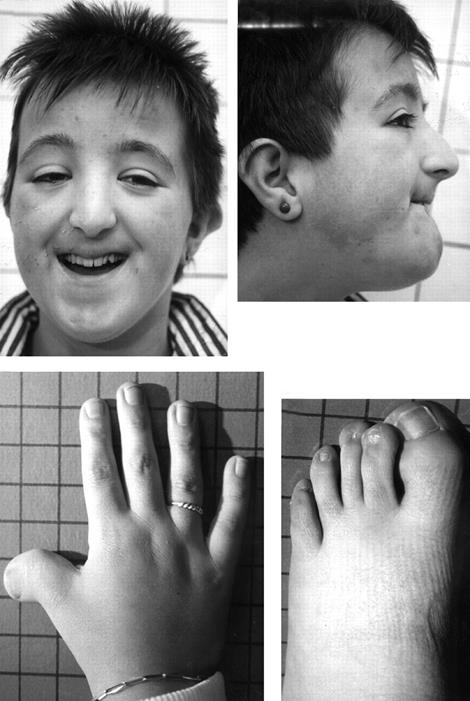
Рисунок XI, 9.Синдром Рубинштейна-Тейби. По материалам сайта http://bjo.bmj.com/content/84/10/1177/F1.large.jpg.
11.3 Генетика эмоционально-личностных расстройств и девиантного поведения
Шизофрения – группа прогредиентных психических заболеваний, протекающих с характерными изменениями личности - эмоциональное оскудение, утрата единства, потеря связи с реальностью, расстройства мышления и развитие бредовых, кататонических и аффективных расстройств. Описано 14 различных локусов, связанных с предрасположенностью к заболеваниям подобного рода, в районах 5q33-q35, 11q14-q21, 6p23, 22q11, 6q13-q26, 8p22-p21, 13q32, 18p, 1q42, 15q15, 10q22, 1p36.2, 15q13.3, 2q32.1.
Ранний детский аутизм - нарушение развития нервной системы, для которого характерно наличие триады: недостаток социальных взаимодействий, нарушенная взаимная коммуникация, ограниченность интересов и повторяющийся репертуар поведения. Это расстройство возникает в результате нарушения развития головного мозга и характеризуется выраженным и всесторонним дефицитом социального взаимодействия и общения, а также ограниченными интересами и повторяющимися действиями. Все указанные признаки проявляются в возрасте до трёх лет. Схожие состояния, при которых отмечаются более мягкие признаки и симптомы, относят к расстройствам аутистического спектра. Заболевание отличается генетической гетерогенностью – известно 17 аутосомных - 7q22, 7q11, 13q14, 15q11, 2q, 17q11, 17q21, 3q25-q27, 7q31, 7q36, 1q41, 21p13-q11, 12q14,16p11.2, 7q35-q36, 3q24, 11q13 - и три X-сцепленных локуса предрасположенности к аутизму. В некоторых случаях известны мутации определенных генов, связанные с этим заболеванием. Например, три X-сцепленные формы этого заболевания обусловлены мутациями в генах NLGN3 (HSAXq13), NLGN4 (HSAXp22.33) и MECP2 (HSAXq28), которые кодируют нейролигин 3, нейролигин 4 и метил-CpG-связывающий белок 2, соответственно. Три аутосомные формы – AUTS15, AUTS16 и AUTS17 – вызваны мутациями в генах CNTNAP2 (HSA7q35-q36), SLC9A9 (HSA3q24) и SHANK2 (HSA11q13).
Синдром дефицита внимания и гиперактивности (СДВГ) - неврологическо-поведенческое расстройство развития, которое начинается в детском возрасте и проявляется в трудности концентрации внимания, гиперактивности и плохо управляемой импульсивности. СДВГ можно диагностировать, начиная с позднего дошкольного или школьного возраста, так как для постановки диагноза необходима оценка поведения ребенка как минимум в двух условиях обстановки (например, дома и в школе). Наличие нарушений обучения и социальных функций является необходимым критерием для установления диагноза СДВГ. Одним из главных признаков СДВГ, наряду с нарушениями внимания, является импульсивность — недостаток контроля поведения в ответ на конкретные требования. Такие дети быстро реагируют на ситуации, не дожидаясь указаний и инструкций, которые позволяют выполнить задание, и неадекватно оценивают требования задания. Они очень небрежны, невнимательны, беспечны и легкомысленны, не всегда могут рассмотреть потенциально негативные последствия своих поступков. Показана связь СДВГ с мутациями в восьми генах: DRD5 (HSA4p16), DAT1 (HSA5p15), HTR1B (HSA6q1), ADRA2A (HSA10q24), DRD4 (HSA11p15), SCN8A (HSA12q13), SNAP25 (HSA20p11.2) и COMT (HSA22q11).
Предрасположенность к алкоголизму в первую очередь обусловлена мутантными аллелями генов кластера ADH (HSA4q22), кодирующих различные субъединицы алкогольдегидрогеназы. Этот фермент катализирует окисление спиртов до альдегидов и кетонов. При его дефиците, этанол накапливается в организме, что усиливает токсическое действие алкоголя. Известно, что в районах традиционного употребления виноградного вина (например, Средиземноморье, Кавказ) наблюдается наименьшая частота встречаемости аллелей, кодирующих дефектную алкогольдегидрогеназу. Высокая частота мутантных аллелей наблюдается в Скандинавии, среди индейцев и азиатских монголоидов, что во многом определяет большее распространение алкоголизма у этих народов.
Кроме генов ADH, на развитие алкогольной зависимости влияют гены SNCA (HSA4q22.1), GABRA2 (HSA5q34), NPY (HSA7p15), TAS2R16 (HSA7q31), TAS2R38 (HSA7q35), CHRM2 (HSA7q35), ANKK1 (HSA11q23), DRD2 (HSA11q23), ALDH2 (HSA12q24), NRXN3 (HSA14q), SLC6A4 (HSA17q) и гены опиоидных рецепторов.
Криминальное поведение характерно для лиц, страдающих некоторыми хромосомными болезнями (например, синдром Клайнфельтера, синдром дополнительной Y-хромосомы – раздел 5.8).
Кроме того, было показано на большой выборке (более 14000) приемных детей из числа осужденных наличие корреляций (статистических связей) по криминальному поведению с биологическими родителями и отсутствие таковой с приемными родителями. Это свидетельствует о значительном влиянии наследственных факторов в склонности к совершению преступлений.
12.4 Наследственные формы нарушений опорно-двигательного аппарата.
Прежде всего, следует отметить, что микро- и макроцефалия, деформация грудной клетки и позвоночника, шеи и другие аномалии опорно-двигательного аппарата характерны для многих хромосомных болезней (разделы 5.8 и 5.9).
Группа заболеваний – дисплазия соединительной ткани (ДСТ) (синонимы: гипермобильный синдром, наследственная коллагенопатия, врожденная соединительнотканная недостаточность) – насчитывает около 200 генетически гетерогенных и клинически полиморфных патологических состояний, обусловленных нарушением развития соединительной ткани в эмбриональном и постнатальном периодах. Для этой группы характерны дефекты волокнистых структур и основного вещества соединительной ткани, которые приводят к различным морфофункциональным нарушениям висцеральных и локомоторных органов с прогредиентным течением. В основе этих заболеваний лежат мутации генов, прямо или опосредованно ответственных за синтез и пространственную организацию коллагена.
Выделяют дифференцированную и недифференцированную дисплазию соединительной ткани. Дифференцированная ДСТ включает в себя синдромы несовершенного остеогенеза, Элерса-Данлоса, Марфана, Стиклера, эластической псевдоксантомы, гаргоилизма и др. Недифференцированная ДСТ - определяющий вариант ДСТ с клиническими проявлениями, не укладывающимися в структуру наследственных синдромов (объединенных под общим названием «гипермобильность»).
Несовершенный остеогенез (НО) – группа аутосомно-доминантных заболеваний, связанных с недостатком коллагена или с его качественным несовершенством (Рисунок XI, 10). Поскольку коллаген является основным белком костной ткани, для этой группы заболеваний характерна ломкость и деформации костей. Известно 8 типов НО с разной степенью поражения костной и соединительной ткани, большинство из которых вызваны различными мутациями в генах α-1- и α-2-цепей коллагена первого типа - COL1A1 (HSA 17q21.31-q22) и COL1A2 (HSA 7q22.1).
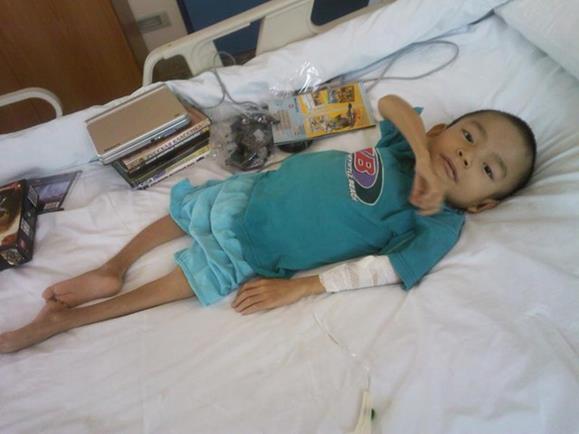
Рисунок XI, 10.Несовершенный остеогенез.http://forum.materinstvo.ru/index.php?showtopic=236470
Синдром Марфана – описан в разделе 9.3.
Синдром Элерса-Данлоса (гиперэластичность кожи, несовершенный десмогенез, несовершенный десмогенез Русакова) – группа заболеваний, вызванных повреждением или недостатком коллагена типов I, III и V. При этих болезнях повреждаются суставы, кожа и кровеносные сосуды. Характерны свободные, сильно гнущиеся суставы, гладкая или эластичная, легко повреждающаяся кожа, неправильное заживление ран и формирование шрамов, маленькие и хрупкие кровеносные сосуды. Все типы синдрома затрагивают суставы, вызывая их гиперподвижность, что позволяет больным выходить за пределы нормального диапазона движений (Рисунок XI, 11).


Рисунок XI, 11.Гиперподвижность суставов при синдроме Элерса-Данлоса. По материалам сайта http://ru.wikipedia.org/wiki/Синдром_Элерса_—_Данлоса.
Типы I и II (классические) – аутосомно-доминантные заболевания соединительной ткани, для которых характерна гладкая, сильно эластичная, легкоранимая кожа, уродливые или необычайно обширные шрамы, гиперподвижные суставы, тенденция к вывихам, склонность к развитию грыжи или смешению любого внутреннего органа. Молекулярно-генетической основой этих типов синдрома являются мутации в генах COL5A1 (HSA 9q34.2-q34.3), COL5A2 (HSA2q31) и COL1A1 (HSA 17q21.31-q22).
Тип III (гиперподвижность) имеет аутосомно-доминантный тип наследования, проявляется в наличии свободных, нестабильных суставов, плоскостопии, высоком и узком нёбе, раннем начале остеопороза, поражениях сердца. Причиной являются мутации в гене COL3A1 (HSA 2q31).
Тип IV (сосудистый) наследуется по аутосомно-доминантному типу. Для него кроме общих для синдрома симптомов характерна хрупкость стенок сосудов оболочек органов и нежной кожи, склонность к разрывам и образованию аневризмы. Так же как и в случае гиперподвижности этиологической причиной являются мутации в гене COL3A1 (HSA 2q31).
Остальные типы синдрома Элерса-Данлоса встречаются достаточно редко, наследуются по аутосомно-доминантному, аутосомно-рецессивному или X-сцепленному рецессивному типу, могут быть связаны с мутациями в генах цепей различных коллагенов или ферментов, участвующих в синтезе коллагена и сборке коллагеновых волокон.
Гаргоилизм (мукополисахаридозы) – описан в разделе 9.3.
Аходроплазия – аутосомно-доминантная карликовость из-за недоразвития длинных костей. Средний рост больных мужчин – 131 см, больных женщин – 123 см. При ахондроплазии отмечается существенное укорочение конечностей при нормальной длине тела, относительная макроцефалия, короткие пальцы, седловидный нос (Рисунок XI, 12). Интеллект, как правило, не поврежден. Молекулярно-генетической причиной этого заболевания являются мутации в гене FGFR3 (HSA4p16.3), который кодирует рецептор-3 фактора роста фибробластов.

Рисунок XI, 12. Пример аходроплазии – Джейсон Акуна, телеведущий. По материалам сайта http://en.wikipedia.org/wiki/File:Jason_Acuña_-_Wee-Man_-_Waterfront_Marriott,_Portland,_Oregon_-_August_15,_2009_-_Full_Body.jpg
Миотоническая дистрофия (болезнь Шнайнерта) – многосистемное наследственное заболевание, основным симптомом которого является запаздывание расслабления мышц после их сокращения. Это аутосомно-доминантное заболевание с манифестацией в возрасте 20-40 лет. Первые признаки заболевания отмечаются в мышцах лица, шеи, рук и ног. Отмечаются опущение век глаз (птоз), слабость мышц лица и речедвигательные нарушения. Наряду с этим характерны катаракты, гипогонадизм, кисты яичника, прогрессирующая умственная отсталость. Болезнь вызвана экспансией тринуклеотидных повторов ЦТГ 3’-фланкирующем районе гена протеинкиназы миотонической дистрофии DMPK (HSA19q13.2-q13.3).
Дистрофия Дюшенна – сцепленная с полом рецессивная прогрессирующая мышечная дистрофия. Первые признаки заболевания проявляются в возрасте 1-3 лет слабостью мышц тазового пояса. Для этого заболевания характерны симметричная атрофия мышц в сочетании с сердечно-сосудистыми, костно-суставными и психическими нарушениями (Рисунок XI, 13). Костно-суставные нарушения при дистрофии Дюшенна характеризуются деформациями позвоночника, стоп, грудины. Этиологическая причина заболевания – различные мутации в гене DMD (HSAXp21.2), который кодирует аподистрофин 1.
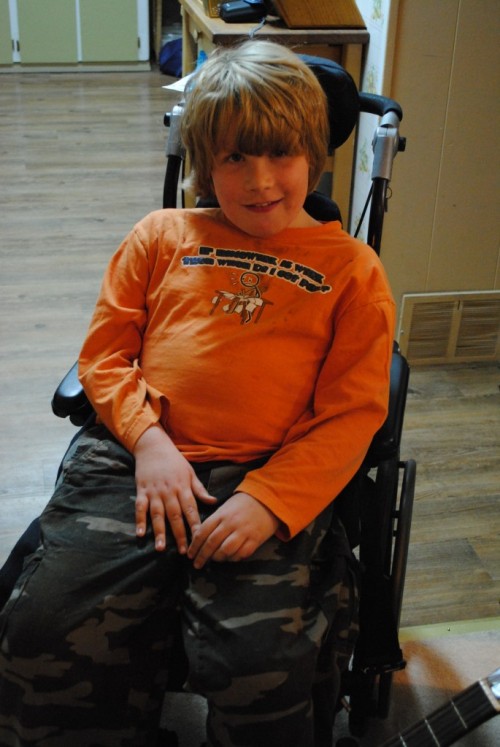
Рисунок XI, 13.Миодистрофия Дюшенна. По материалам сайта http://www.growingyourbaby.com/2010/09/08/tutus-for-tanner/nikon-2010-071-685x1024/
11.5 Наследственные формы глухоты и тугоухости в детском возрасте
Микротия с атрезией наружного слухового прохода и проводящей тугоухостью – наследственное заболевание с невыясненной генетической природой, для которого характерны отсутствие или существенная деформация ушной раковины и отсутствие просвета слухового канала. Показано достоверное различие конкордантности по этому признаку у однояйцовых и разнояйцовых близнецов.
Синдром Ушера – аутосомно-рецессивное заболевание, которое проявляется во врожденных нарушениях слуха различной степени, прогрессирующей пигментной дегенерации сетчатки (Рисунок XI, 14), приводящей к постепенному сужению полей зрения и слепоте, и, иногда, потере чувства равновесия. В мире 5-6% от общего числа людей с любыми нарушениями слуха и 50% слепоглухих страдают этим синдромом. Клинический полиморфизм определяется генетической гетерогенностью – известно 12 локусов, связанных с разными типами этого заболевания. Наиболее известна форма синдрома Ушера, вызваная мутациями гена MYO7A (HSA11q13.5), который кодирует миозин 7А.
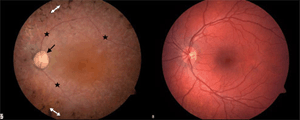
Рисунок XI, 14.Фотография сетчатки при синдроме Ушера (слева) и в норме (справа). Зрительный нерв (обозначен черной стрелкой) бледный, сосуды тонкие (обозначены звездочками), присутствуют пигментные гранулы (обозначены белыми двойными стрелками). По материалам сайта http://my.clevelandclinic.org/disorders/usher_syndrome/hic_usher_syndrome.aspx.
Синдром Крузона (черепно-лицевой дизостоз) – аутосомно-рецессивное заболевание, вызванное преждевременным срастанием венечного и сагитального швов черепной коробки. Для синдрома характерно западание глазниц и костей щек, выпученные глаза, вдавленное переносье, выступающая челюсть (Рисунок XI, 15).У большинства больных отмечаются нарушения слуха. Фенотип, свойственный этому синдрому, развивается по причине мутаций в гене FGFR2 (HSA10q26), кодирующем рецептор 2 фактора роста фибробластов.
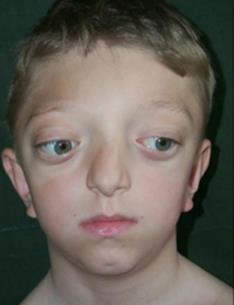
Рисунок XI, 15.Синдром Крузона.По материалам сайта http://childface.ru/rus/content/36/diagnosticheskaya_baza.html.
Синдром Тричера-Коллинза – аутосомно-доминантное заболевание с характерными изменениями лицевого черепа – небольшими размерами рта, подбородка, ушей, - косоглазием и колобомой век (Рисунок XI, 16). Характерны аномалии наружного слухового прохода и тугоухость. Этиологической причиной являются мутации в гене TCOF1 (HSA5q31.3-q33.3), который кодирует ядерный белок с LIS1-гомологичным доменом.
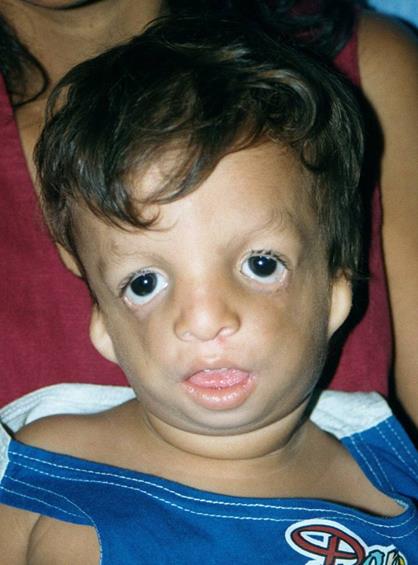
Рисунок XI, 16. Синдром Тричера-Коллинза. По материалам сайта http://emedicine.medscape.com/article/946143-overview.
Синдром Альпорта – сочетание наследственного нефрита и глухоты (Рисунок XI, 17). Симптоматика со стороны выделительной системы появляется в 5-10 лет. Постепенная потеря слуха обычно начинается раньше. Вначале это нейросенсорное снижение слуха высоких тонов, затем - низких, переходящее из звукопроводящей в звуковоспринимающую тугоухость. Иногда наблюдается миастения, потеря памяти и интеллекта. Сцепленная с полом рецессивная форма этого синдрома вызвана мутациями в гене α-5 цепи коллагена типа IV – COL4A5 (HSAXq22.3). Аутосомно-рецессивная форма синдрома Альпорта связана с мутациями в генах COL4A3 и COL4A4 (HSA2q36-q37), которые кодируют α-3 и α-4 цепи коллагена типа IV. Генетическая природа аутосомно-доминантной формы пока изучена меньше, но предполагается ее связь с мутациями гена COL4A3.
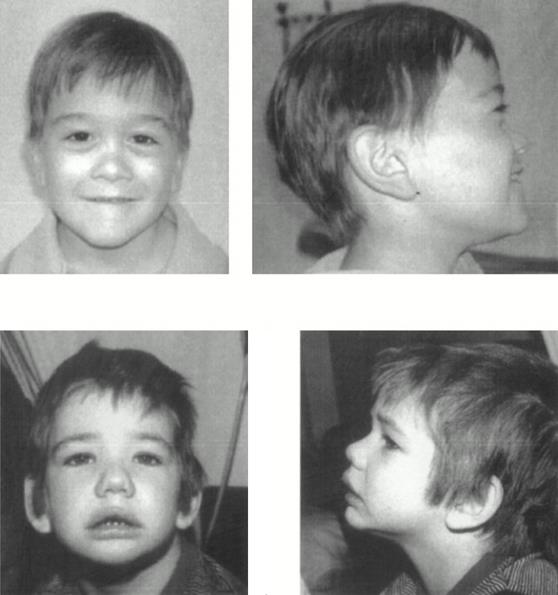
Рисунок XI, 17. Синдром Альпорта. По материалам сайта http://jmg.bmj.com/content/39/5/359/F1.large.jpg
Синдром Пендреда – аутосомно-рецессивное заболевание, проявляющееся потерей слуха и патологиями щитовидной железы – узловым зобом и гипотериозом. Для детей с этим заболеванием характерна прогрессирующая потеря слуха, которая начинается обычно в возрасте до трех лет, замедленный рост костей, диспропорции скелета – большой череп, относительно короткие конечности (Рисунок XI, 18). Генетической основой заболевания являются мутации в гене PDS (HSA7q31), который кодирует пендрин – фермент, присутствующий в улитке внутреннего уха, щитовидной железе и почках.
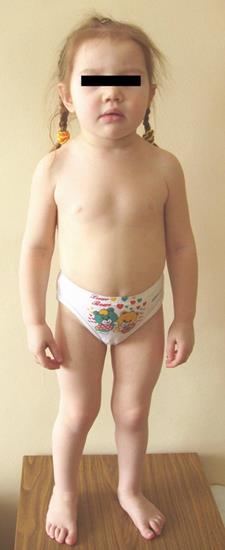
Рисунок XI, 18.Синдром Пендреда. Девочке 6 лет. Длина тела 92 см (соответствует 3-летнему возрасту). По материалам сайтаhttp://www.medico.ru/medatlas/post/1115312770_01_168_1.jpg
Синдром Ричардса-Рандля – аутосомно-рецессивное заболевание, проявляющееся в атаксии, гипогонадизме и нейросенсорной глухоте. У больных развивается глубокое слабоумие, прогрессирующие мышечные атрофии, наблюдается гипоплазия половых желез, недоразвитие вторичных половых признаков, кетоацидурия, прогрессирующая нейросенсорная тугоухость с раннего детства. Молекулярно-генетические основы этого заболевания остаются неизвестными.
Синдром Жервелла и Ланге-Нильсена – аутосомно-рецессивное заболевание, которое характеризуется врожденной глухотой и нарушением ритма сердца – удлиненным интервалом QT, отражающего процессы электрического возбуждения и восстановления сердечной мышцы. Часто у больных отмечаются множественные пороки сердца, имеется высокий риск синкопе и внезапной смерти. Клинический полиморфизм, проявляющийся в широком варьировании тяжести проявления от мутациями в гене KCNQ1 (HSA11q15.5), который кодирует один из белков калиевых каналов.
Синдром множественных лентиго (синдром леопарда) – аутосомно-доминантное заболевание, минимальные диагностические признаки которого: множественные лентиго (пигментные пятна размером 1-5 мм более темные, чем веснушки), стеноз легочной артерии, умеренный гипертелоризм, аномалии гениталий, глухота (Рисунок XI, 19). Этиологической причиной являются мутации в гене PTPN1 (HSA12q24.1), кодирующего протеин-тирозин-фосфатазу нерецепторного типа 11.
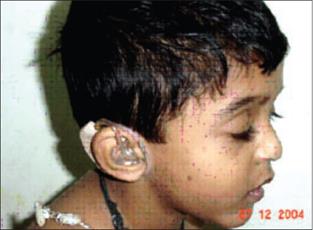
Рисунок XI, 19.Синдром множественных лентиго. По материалам сайта http://www.gfmer.ch/genetic_diseases_v2/gendis_detail_list.php?offset=15&cat3=177.
Синдром Ваарденбурга – наследственное заболевание, имеющее следующие признаки: телекант (латеральное смещение внутреннего угла глаза), гетерохромия радужной оболочки, седая прядь надо лбом и врожденная глухота (Рисунок XI, 20). Глухота вызвана нарушениями спирального (кортиева) органа с атрофическими изменениями в спинальном узле и слуховом нерве. Лечение глухоты при этом синдроме неэффективно. Синдром Ваарденбурга типов 1 и 3 вызваны доминантными мутациями в гене PAX3 (HSA2q35), который кодирует транскрипционный фактор семейства PAX. Пенетрантность этих мутаций неполная, экспрессивность варьирует.
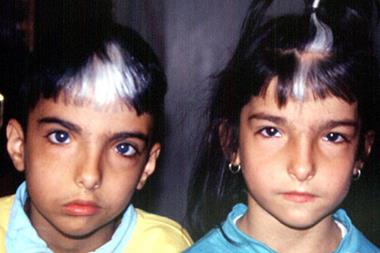
Рисунок XI, 20. Синдром Ваарденбурга. По материалам сайта http://emedicine.medscape.com/article/950277-overview.
11.6 Генетически обусловленные формы детской слепоты и слабовидения
Слепота и слабовидение наблюдаются при синдромах Марфана (раздел 9.3), Альпорта, Крузона, Ушера (раздел 12.5), о которых речь шла выше.
Синдром Ригера (открытоугольная ювенильная глаукома) – аутосомно-доминантное заболевание, которое проявляется в повышении внутриглазного давления, развитии оптической нейропатии с последующей атрофией головки зрительного нерва и возникновением дефектов поля зрения. Для больных характерна щелевидная деформация зрачка, эктопия хрусталика, колобома сосудистой оболочки глаза, телекант, аномалии зубов (Рисунок XI, 21). Этиологической причиной этого генетически гетерогенного заболевания могут быть мутации в генах PITX2 (HSA4q25-q26), FOXC1 (HSA6p25) и делеция района HSA13q24.
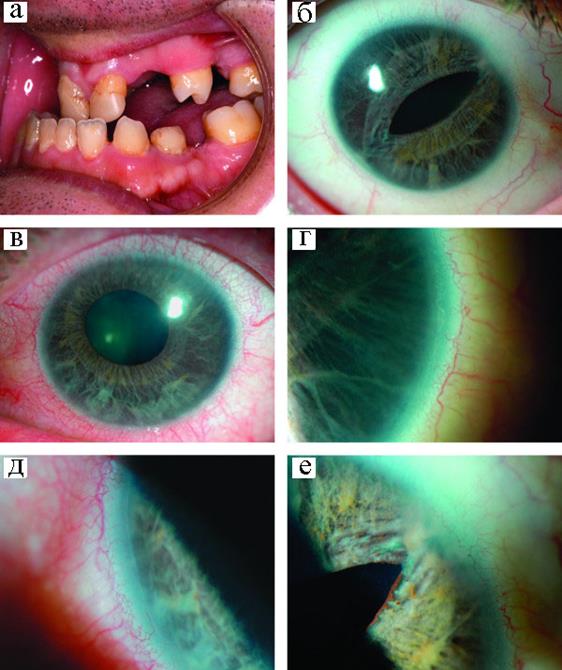
Рисунок XI, 21. Синдром Ригера. А – микродонтия и гиподонтия, Б – щелевидный зрачок и атрофия радужной оболочки правого глаза, В – смещение зрачка с атрофией радужной оболочки левого глаза, Г – задний эмбриотксон правого глаза, Д - задний эмбриотоксон левого глаза, Е – периферическая предняя синехия правого глаза. По материалам сайта http://www.casesjournal.com/content/1/1/299
Синдром Альстрема характеризуется пигментной дегенерацией сетчатки, ожирением, сахарным диабетом, дилятационной кардиомиопатией, нефропатией и прогрессирующей нейросенсорной глухотой (Рисунок XI, 22). Дети при рождении имеют нормальную массу тела, но к концу первого года жизни появляется ожирение. У больных развивается нистагм (дрожание глазных яблок), прогрессирует дистрофия нейроэпителия с атрофией и пигментной инфильтрацией внутренних пластов сетчатки. К 7 годам часто происходит полная потеря зрения. Для заболевания характерен клинический полиморфизм, который наблюдается даже у сибсов. Мутации в гене ALMS1 (HSA2p13), продукт которого пока не охарактеризован, являются причиной этого заболевания.
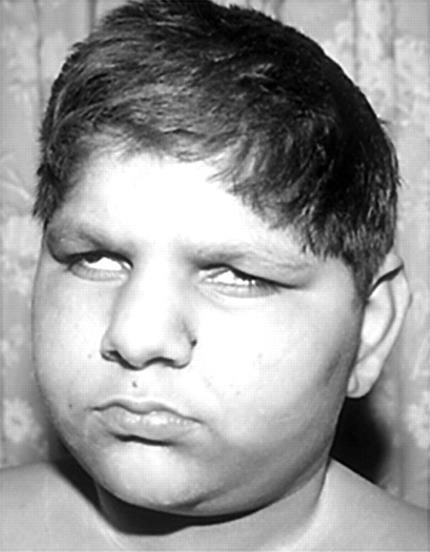
Рисунок XI, 22. Синдром Альстрема. По материалам сайта http://jmg.bmj.com/content/42/2/e10.extract
Синдром Ленца – сцепленное с полом рецессивное заболевание, которое проявляется в нарушении развития глаза (микрофтальмия или анофтальмия), связанных со снижением остроты зрения или слепотой. Для заболевания также характерны аномалии развития ушей, зубов, скелета, выделительной и половой систем (Рисунок XI, 23). Извеcтна локализация гена, мутация которого приводит к этому заболеванию, в районе HSA Xq27-q28. Кроме синдрома Ленца описано около десятка форм изолированной и синдромной микрофтальмии с аутосомно-доминантным, аутосомно-рецессивным и сцепленным солом наследованием.
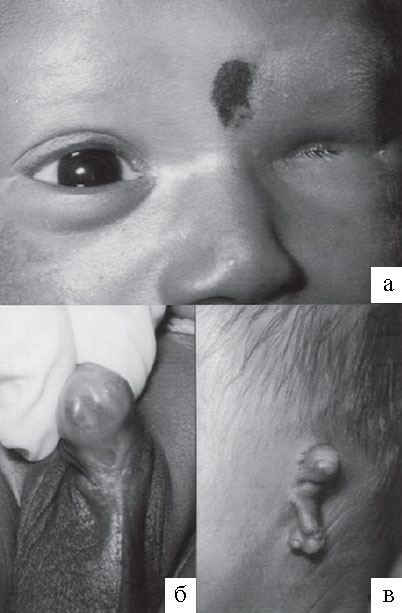
Рисунок XI, 23. Синдром Ленца. А - микрофтальмия, Б – односторонний крипторхизм, В - микротия. По материалам сайта http://www.gfmer.ch/genetic_diseases_v2/gendis_detail_list.php?cat3=1883
Наследственные катаракты – группа заболеваний, связанных с помутнением хрусталика глаза и вызывающее различные степени расстройства зрения. Описано 14 изолированных форм этого заболевания с аутосомно-доминантным, аутосомно-рецессивным и сцепленным с полом типами наследования. Кроме того катаракта в качестве симптома может входить во многие наследственные патологические состояния.
Синдром Апера (Аперта) – заболевание с аутосомно-доминантным типом наследования, которое характеризуется синостозом (срастанием) венечных швов, плоским лбом, гипертелоризмом, антимонголоидным разрезом глаз, плоскими глазными впадинами, запавшей переносицей, полным сращением II-V пальцев кистей и стоп, атрофией зрительного нерва (Рисунок XI, 23). Больные отстают в умственном и физическом развитии. Молекулярно-генетической основой заболевания являются две мутации в экзоне 7 гена FGFR2 (HSA10q26), который кодирует рецептор 2 факторов роста фибробластов.

Рисунок XI, 23.Синдром Апера.По материалам сайта http://www.examiner.com/special-education-in-los-angeles/apert-syndrome-information-and-activities-for-early-childhood
Синдром Маршалла - аутосомно-доминантное заболевание, характеризующееся дисплазией лица (нос курносый седловидной формы), гипоплазией средней части лица, которая создает впечатление лица бульдога (Рисунок XI, 24), гиперплазией надглазничной области, аномалиями зубов (адентией, гиподонтией, удвоением зубов, микродентией), аномалиями глаз — врожденной близорукостью и быстро прогрессирующей катарактой. У больных наблюдается врожденная, часто прогрессирующая односторонняя или двусторонняя тугоухость, которая позже нередко переходит в полную глухоту. Этиологической причиной заболевания являются мутации в гене COL11A1 (HSA1q21), который кодирует α-1 цепь коллагена типа XI.

Рисунок XI, 24. Синдром Маршалла. По материалам сайта http://www.gfmer.ch/genetic_diseases_v2/gendis_detail_list.php?cat3=703
Синдром Дюэйна - Аутосомно-доминантное заболевание, для которого характерны односторонняя слабость латеральной прямой мышцы глаза (уменьшение или отсутствие способности глаза к отведению, ограничено приведение и слабая конвергенция, ретракция глазного яблока при приведении) с сужением глазной щели во время приведения и расширением ее во время отведения (Рисунок XI, 25). Иногда у больных наблюдаются аномалии развития лица, зубов, ушей.
Для этого заболевания характерна генетическая гетерогенность – синдромы Дюэйна I и II типов вызваны мутациями локуса DURS1 (HSA8q13) и гена CHN1 (HSA2q31), который кодирует α-1 химерин.

Рисунок XI, 25. Синдром Дюэйна. По материалам сайта http://emedicine.medscape.com/article/1198559-overview.
Наследственный дальтонизм – генетически обусловленная неспособность различать один или несколько цветов. Полное отсутствие цветового зрения – достаточно редкое явление. Наиболее часто встречается пронатопия – дефект рецепторов, воспринимающих свет в красной области спектра. Это заболевание вызвано мутациями в гене OPN1LW (HSA Xq28), который кодирует чувствительный к свету длинных волн опсин 1. Другие мутации того же гена приводят к развитию дейтеранопии – заболевания, при котором смешиваются красный и зеленый цвета.
Контрольные вопросы и задания к главе XI
1. Выберите правильный ответ:
Дисплазия соединительной ткани как правило связана с дефектами
а) коллагена
б) гликогена
в) гемоглобина
2. Для каких заболеваний характерны одновременно нарушения зрения и слуха?
3. Есть ли связь между слабоумием и криминальным поведением? Если есть, при каких заболеваниях?
4. Существуют ли географические закономерности предрасположенности к алкоголизму?
5. Для каких синдромов показано влияние геномного импринтинга?
Дополнительная литература к главе XI
Н.П. Бочков. Клиническая генетика // М.: Гэотар-Мед. 2002. – 457 С.
Дата добавления: 2015-07-22; просмотров: 8161;