Реакция Вюрца. Аллильное сочетание и родственные случаи
Выше мы уже обсуждали реакцию Вюрца как один из простейших случаев образования связи С—С. В этой реакции одна молекула алкилгалогенида выступает в роли элекгрофила (эквивалента карбокатиона), в то время как вторая под действием металла превращается в соответствующий алкилметал, который исполняет роль нуклеофильной компоненты сочетания (эквивалента карбаниона). Отмечалось также, что эта давно известная реакция была модифицирована (за счет изменения природы нуклеофильной компоненты, т.е. перехода к использованию купратных реагентов) таким образом, что в настоящее время сочетание по схеме реакции Вюрца может считаться действительно общим методом синтеза.
С учетом сказанного легко спланировать модельный синтез, скажем я-бу-тана, соответствующий какой-либо из альтернативных схем разборки по ге-теролитическому механизму:
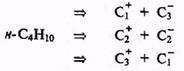
Все эти три схемы в общем равноценны и вполне реальны, так как требуемые электрофилы, например СН3I, С2Н5I и С3Н7I, и нуклеофилы, например CH3MgI, C2H5MgI и C3H7MgI (модифицированные солями меди), легко доступны. Можно, конечно, возразить, что синтезировать н-бутан вообще не нужно, так как этот углеводород в изобилии поставляется нефте- и газоперерабатывающей промышленностью. Тем не менее, эта задача может реально возникнуть, если для исследовательских целей неободимо иметь н-бутан, содержащий метку (например, дейтерий или 13С) в заданном положении. В этом случае выбор подходящей пары электрофил+нуклеофил будет определяться более всего доступностью соответствующих меченых предшественников.
Однако в синтезе чаще встречаются задачи, в которых формально возможные альтернативы на самом деле не являются равноценными. Рассмотрим, например, синтез углеводорода 50 (схема 2.17). В этой молекуле имеют
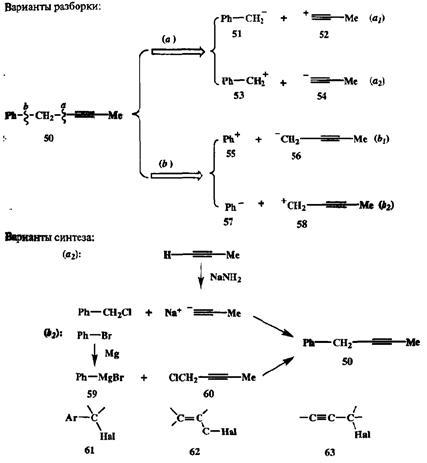 Схема 2.17
Схема 2.17
|
ся два легко распознаваемых структурных элемента, которые входят в состав многих легкодоступных соединений, а именно: бензольное ядро и остаток метилацетилена. Очевидно, что в синтезе 50 разумно использовать исходные вещества, уже содержащие эти фрагменты. Учет таких соображений позволяет предложить два варианта разборки целевой молекулы, показанных на схеме 2.17 (путь «о» и путь «*»).
Разборка по связи бензил — этинил (путь «а») приведет нас к двум парам ионов 51 + 52 или 53 + 54. Для бензил-аниона (51) легко найти синтетический эквивалент в виде бензилмагнийбромида. Однако не имеется очевидного реагента, способного служить эквивалентом этинильного катиона 52, и поэтому можно исключить из дальнейшего рассмотрения вариант *at» как мало перспективный. Напротив, вариант «а2» вполне реален, так как для обоих ионов 53 и 54 имеются хорошо известные эквиваленты в виде бензил-хлорида и натриевого производного метилацетилена соответственно. Примерно такая же ситуация возникает при анализе разборки по связи фенил — пропаргил (путь «6»). Здесь также можно рассматривать варианты сборки этой связи из пар ионов 55 + 56 или 57 + 58.
Для реализации первого из этих вариантов «Z>i» необходимо подобрать синтетический эквивалент для фенил-катиона (55), что отнюдь не просто (для аниона 56 таким эквивалентом служит соответствующий пропаргиллитиевый реагент). Напротив, в варианте «Ьг» нуклеофильный 57 и элсктрофильный 58 компоненты легко идентифицируются в виде соответственно фенилмагний-бромида (59) и пропаргилхлорида (60). Таким образом из четырех формально равноценных вариантов решения задачи синтеза 50 мы приходим к двум — варианты *а2» и «fa», и оба они реально осуществимы (см. схему 2.17).
В реакциях, которые мы только что разбирали, фигурируют очень типичные синтетические эквиваленты органических ионов — это нутслеофилы (реагенты Гринъяра и ацетилениды) и электрофилы (бензил- и пропаргилгало-гениды). Общей чертой этих электрофилов является наличие системы тг-электронов по соседству с потенциальным карбокатионным центром. Это обеспечивает повышенную легкость замещения по этому центру и, поэтому реагенты, содержащие бензильную (61), пропаргильную (63) или аллильную (62) группировки, особенно эффективны как эквиваленты карбокатионов.
Учет этого обстоятельства указывает на то, что в общем случае ретросинтети-ческий анализ целевых структур, содержащих кратные углерод-углеродные связи или ароматические ядра, целесообразно начинать с разборки связи С-С у ал-лильного, пропаргильного или бензильного центров с тем, чтобы выйти к паре бснзил(аллил- или пропаргил-)-катион + карбанион и уже далее анализировать доступность реагентов, необходимых для реализации такой схемы [6].
Проиллюстрируем сказанное на конкретном примере синтеза непредельного спирта 64 — полового аттрактанта яблоневой плодожорки Laspeyresia pomonella, распространенного вредителя яблоневых садов [7] (схема 2.18). Разборка структуры 64 по связи у аллильного атома углерода привела к ал-лильному катиону 65 и функционализованному алкильному аниону 66. Очевидными эквивалентами этих ионов служили бромид 61 и реагент Гриньяра
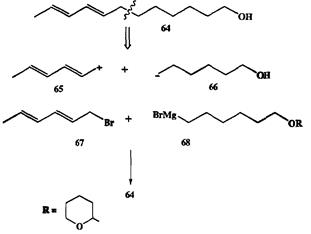 Схема 2.18
Схема 2.18
|
68, содержащий защищенную (в виде дигидропиранильного производного) гидроксильную группу. И электрофил 67, и нуклеофил 68 легко получались из доступных исходных веществ, и их конденсация протекала с хорошим выходом. После этого потребовалось лишь снять защиту, чтобы получить целевой продукт 64.
Подвижная система л>электронов кратных связей С—С, ответственная за стабильность соответствующих карбокатионов, оказывает столь же эффективное действие на стабильность карбанионов. Вследствие этого разборка по аллильной, бензильной или пропаргильным связям выигрышна еще и потому, что получающийся при этом непредельный фрагмент может быть представлен не только как карбокатион, но и как карбанион. Подобный дуализм всегда полезен, поскольку он расширяет область поиска наиболее подходящих вариантов, но особенно часто им пользуются при синтезе целого рада представителей одного из важнейших классов природных соединений, а именно ациклических изопреноидов*.
Структуры многих представители этого класса соединений выглядят так, как будто они были специально предназначены для ретросинтетического анализа с разрывом связи С-С между аллильными атомами углерода.
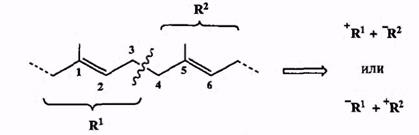 Схема 2.19
Схема 2.19
|
В самом деле, типичные представители этих природных соединений содержат 1,5-диеновую систему, разборка которой по центральной связи С-3—С-4 автоматически приводит к двум аллильным фрагментам (схема 2.19). Любой из них может в принципе рассматриваться как карбокатион или карбанион. Образование связи С-С путем сочетания двух таких фрагментов может определенно считаться «верным делом», и по этой причине во множестве синтезов изопреноидов используются в качестве «строительных блоков» аллильные заготовки самого причудливого строения. Выбор конкретной природы реагентов на этой ключевой стадии определяется, в основном, доступностью соответствующих исходных соединений. При этом, правда, приходится еще считаться с возможностью аллильной перегруппировки как в карбокатионах, так и карбанионах, что накладывает некоторые ограничения и на природу используемых реагентов, и на условия проведения такого сочетания**.
Дата добавления: 2015-04-05; просмотров: 1555;