Карбонильные соединения как нуклеофилы и электрофилы
В определенном смысле карбонилсодержащий фрагмент С-С=О может рассматриваться как аналог аллилъной системы С—С=С. Однако в отличие от последней в карбонильных соединениях эффективная стабилизация не может быть обеспечена для катиона, поскольку в сильнополяризованной карбонильной группе уже имеется частичный положительный заряд на атоме углерода, что делает энергетически невыгодным появление еще одного положительного заряда на соседнем атоме. Напротив, благодаря этой же поляризации соответствующий анион (енолят-анион) проявляет повышенную стабильность.
Как мы уже говорили, в енолят-анионе заряд распределен между кислородом и а-углеродным атомом. Поэтому в общем случае его реакция с элект-рофилом можгт протекать как атака по атому углерода или атому кислорода, давая соответственно а-замещенные карбонильные производные или виниловые эфиры (схема 2.20).
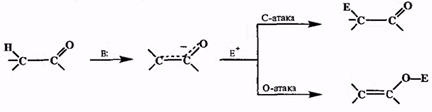 Схема 2.20
Схема 2.20
|
К счастью, направлением атаки электрофила можно управлять, варьируя природу реагента и/или условия проведения реакции. На этом основаны многие методы построения связи С—С, использующие алкилирование енолятов электрофилами. Здесь, однако, встречается одно существенное осложнение, которое заслуживает специального рассмотрения.
Типичные карбонильные соединения ведут себя как очень слабые кислоты. Поэтому если генерировать енолят из ацетона при действии даже такого довольно сильного основания, как этилат натрия, то равновесие этой реакции будет сильно сдвинуто влево (схема 2.21).
 Схема 2.21
Схема 2.21
|
В то же время неионизованные карбонильные соединения являются достаточно активными электрофилами, и поэтому в системе, содержащей и карбонильное соединение, и его енольную форму, т. е. как электрофил, так и нуклеофил, будет легко протекать сочетание этих двух компонент по схеме хорошо знакомой альдольной конденсации. Следовательно, избирательно провести в такой системе алкилирование енолятов каким-либо другим электрофилом практически невозможно.
По этой причине еще сравнительно недавно синтетические возможности использования алкилирования енолятов ограничивались требованием при менения в качестве субстратов реакции особенно стабильных енолятов, получаемых из б-дикарбонильных соединений, например, типа ацетоуксусного или малонового эфиров. Так, классическим методом наращивания углеродной цепи на два звена является реакция алкилгалогенидов с енолятом малонового эфира с последующим декарбоксилированием получаемого алкилмалонового эфира по общей схеме:
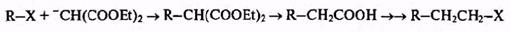
Нетрудно видеть, что в этой последовательности енолят малонового эфира выступает в роли эквивалента енолят-аниона, производного уксусной кислоты.
Подобного рода подход мог вполне успешно применяться и для более сложных случагв, но при этом требовалось предварительно модифицировать структуру монокарбонильного соединения, чтобы создать требуемый (З-ди-карбонильный фрагмент и тем самым гарантировать возможность генерации достаточно стабильного енолята. Необходимость проведения отдельных стадий, связанных с введением и последующим удалением дополнительных функций, несмотря на простоту используемых для этого методов, конечно, создавало определенные неудобства и неоправданно удлиняло всю синтетическую схему. В настоящее время разработан новый подход к генерации енолятов, предполагающий использование сильных оснований, который не требует предварительной модификации карбонильных субстратов, но его целесообразно рассмотреть чуть позже, в связи с проблемой альдольной и родственных ей конденсаций.
Итак, благодаря поляризации карбонильной группы, отражаемой формулой >Ce+=OS", карбонильные соединения можно использовать в виде енолятов как С-нуклеофилы 18]. Легко понять, что та же самая поляризция делает атом углерода карбонильного соединения электрофильным, благодаря чему эти соединения могут также играть роль С-электрофилов — синтетических эквивалентов катиона >C+-OH.
Одна из важнейших реакций с использованием карбонильных соединений как электрофилов — это реакция Гриньяра, присоединение магнийорганиче-ских производных по карбонильной группе. Чистым итогом этого превращения является образование новой связи С-С с одновременным превращением карбонильной функции в спиртовую [9а] (схема 2.22). Общеизвестна широкая применимость этой реакции к самым разным типам карбонильных соединений. Однако также известно, что из-за высокой основности магнийоргани-ческих реагентов в своем классическом виде она неприменима для легко енолизумых производных. Так, например, до недавнего времени совершенно невозможно было провести реакцию Гриньяра с ацетоуксусным эфиром, а с такими кетонами, как показанный на схеме тетралон (69), выход продукта присоединения по карбонильной группе мог оказаться неприемлемо низким.
К счастью, удалось разработать модификацию этой реакции, свободную от такого ограничения: было найдено, что цериевые реагенты (легко получаемые при обработке реактивов Гриньяра стехиометрическим количеством СеCl3) почти лищены основных свойств, и поэтому способны реагировать по обшей схеме синтеза Гриньяра с кетонами практически любых типов (в том числе с 69) независимо от способности последних к енолизации [9Ь]. Даже апетоуксусный эфир ведгт себя «вполне прилично» в реакциях с цериевыии реагентами — просто как обычный кетон!
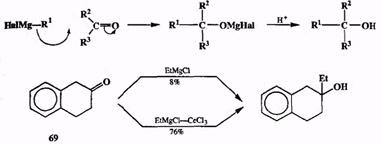 Схема 2.22
Схема 2.22
|
Итак, благодаря общности и надежности реакция Гриньяра может по праву считаться одним из самых эффективных методов создания связи С-С по схеме электрофил + нуклеофил Поэтому стандартным приемом ретросинтетической разборки фрагмента целевой молекулы, содержащего гидроксильную группу, является разрыв связи С-С, примыкающей к гидроксильной функции*.
Ярко выраженный электрофильный характер карбонильной группы делает ее подходящим субстратом для реакций со множеством нуклеофилов, список которых отнюдь не ограничиватся металлоорганическими производными рассмотренных выше типов. Особую важность для синтеза имеют конденсации, в которых по карбонильной группе присоединяются нуклеофилы ишаенолятов (схема 2.23). Спектр превращений этого типа включает такие классические реакции, как альдольная и кротоновые конденсации, реакции Кляйзена, Дарзана, Реформатского, Штоббе, Перкина и Кневенагеля и т. [10]. При довольно значительных различиях в конкретной природе субстратов и условиях проведения ключевая стадия всех этих реакций описывается механизмом, показанном на схеме 2.23.
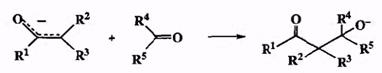 Схема 2.23
Схема 2.23
|
Оба участника такой конденсции — и электрофил, и нуклеофил — являются карбонильными производными: одно — в виде енолята, а другое — в не-ионизованном виде. Понятно, что лишь в том случае, если оба исходных соединения тождественны, не имеет значения, какая молекула будет выступать в роли электрофила, а какая — в роли нуклеофила. Пример такой ситуации — синтез ацетоуксусного эфира из этилацетата (схема 2.24, сложноэфирная конденсация, она же реакция Кляйзена).
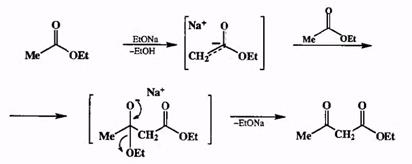 Схема 2.24
Схема 2.24
|
Если же структуры двух карбонильных компонент различны и оба соединения способны к енолизации, то в общем случае можно ожидать образования четырех продуктов. Именно таким будет результат сложноэфирной конденсации этилацетата и этилпропионата, если проводить ее в классических условиях (действием этилата натрия на смесь этих двух эфиров).
Отсюда ясно, что препаративная ценность конденсаций рассматриваемого типа целиком зависит от того, насколько четко удастся распределить роли: кому быть электрофилом, а кому — нуклеофилом.
В классических вариантах конденсации карбонильных производных разделение ролей достигалось с помощью общего приема, а именно использованием в качестве субстратов реакции соединений, резко отличающихся по своей способности к енолизации. Действительно, по указанному признаку, т. е. по природе субстратов и соответственно области применения, более всего отличаются друг от друга названные выше именные реакции. Скажем, в реакции Перкина — конденсации ароматических альдегидов с ангидридами алифатических карбоновых кислот — «игра» построена на том, что в элект-рофильном компоненте (альдегиде) не содержится а-водородов, что вообще лишает его способности образовывать енсляты. В то же время во второй компоненте, используемой как источник нуклеофила (енолята), такой, как, например, уксусный ангидрид, сильно понижена (в сравнении с альдегидом) реакционноспособность карбонильной функции по отношению к карбанионамтипаенолятов, в силу чего не наблюдается самоконденсации с участием этого реагента. В результате реакция протекает вполне однозначно без образования продуктов кросс-сочетания и приводит с хорошим выходом к образованию производных коричной кислоты.
АгСНО + (СН3СО)2О → АгСН(ОН)-СН2СООАс → АгСН=СНСООН
Однако в настоящее время существует гораздо более общее и эффективное решения проблемы «распределения ролей» в таких реакциях. Произошло это прежде всего благодаря разработке нового поколения сильных и ненуклео-фильных оснований, таких, например, как диизопропиламид лития (LDA), которые позволяют генерировать еноляты практически из любых карбонильных производных, содержащих хотя бы один атом водорода в а-положении, в очень мягких условиях и с полным смещением равновесия енолизации вправо.
Подобная методика открыла возможность селективного и полного превращения одной из карбонильных компоненте енолят в условиях, которые в большинстве случаев исключали возможность самоконденсации. Полученный же таким образом енолят мог далее вводиться в реакцию с добавляемым извне карбонильным соединением (или любым другим электрофилом), ено-лкзация которого в этих условиях уже не могла иметь места (из-за израсходования сильного основания). Подобное разделение стадий генерации карба-ниона и его реакции с электрофильным партнером (см. схему 2.25) позволило в широких пределах и независимо варьировать природу обеих компонент конденсации карбонильных производных, не опасаясь того, что в какой-то момент партнеры «спутают свои роли» {4, 10].
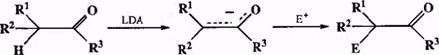 Схема 2.25
Схема 2.25
|
Именно таким образом удается, например, достаточно селективно провести сложноэфирную кросс-конденсацию эфиров уксусной и пропионовой кислоты в обоих вариантах:
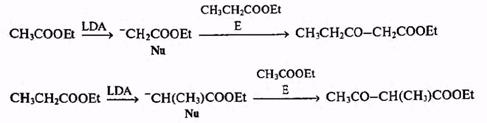
Разработка совершенных и общих методов генерации стабильных еноля-тов самой различной структуры по сути дела нивелировала существовавшие ранее индивидуальные особенности классических реакций конденсации карбонильных соединений. Поэтому, строго говоря, сейчас уже нет логических оснований для классификации этих методов в традиционных терминах именных реакций (Кляйзена, Реформатского, Перкина и др.) и сохранение этих названий — это просто вполне справедливое признание выдающихся заслуг наших великих предшественников.
Нетрудно заметить, что результатом рассмотренных конденсаций является образование продуктов, содержащих кислородсодержащие заместители в положении 1,3. Это могут быть р-гидроксикарбонильные соединения (если оба компонента альдегиды или кетоны), или р-дикарбонилъные для случая сложноэфирной конденсации. Оба этих типа производных обладают высокой и разнообразной реакционной способностью, что значительно расширяет синтетическую значимость методов их получения. Типичные возможности таких превращений, показанные на схеме 2.26 для продукта конденсации ацетона и уксусного альдегида, альдоля 70, это окисление с образованием р-дикетона 71, восстановление, приводящее к 1,3-диолу 72, и дегидратация, дающая а,р-непредельный кетон 73.
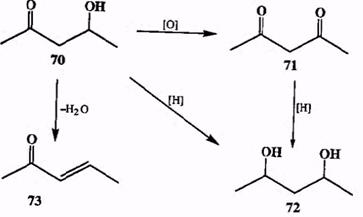 Схема 2.26
Схема 2.26
|
Благодаря надежности и прекрасной отработанности методик проведения указанных превращений мы с полным правом можем рассматривать все четыре типа производных 70—73 как продукты конденсации двух карбонильных производных по типу нуклеофил + электрофил. Отсюда непосредственно следует одна из наиболее очевидных рекомендаций ретросинтетического анализа: если в целевой молекуле содержится фрагмент одного из указанных структурных типов, то следует рассмотреть варианты разборки одной или другой из двух связей между 1,3-атомами углерода, несущими кислородсодержащие заместители. Анализ этих вариантов позволит далее выйти к структурам карбонильных предшественников — кандидатов на роли электрофила и нуклеофила в альтернативных вариантах соответствующей карбонильной конденсации (сейчас все такие конденсации принято называть «альдольными реакциями»).
Ретросинтетический анализ производного 74, фрагмента структуры молекулы сложного макролидного антибиотика 6-дезоксиэритронолида В и полупродукта в синтезе последнего, может служить хорошим примером того, насколько конструктивным может быть применение этого простого принципа (схема 2.27) {11].
В показанном фрагменте содержится набор пар 1,3-кислородсодержаших заместителей, что в соответствии со сказанным выше почти автоматически Диктует ретроальдольную разборку по связям С-С внутри этих фрагментов. Последовательность таких ретросинтетических шагов, показанная на схеме, приводит к быстрому упрощению структур предшественников и в конце концов позволяет придти к простейшим исходным веществам (одна из завершающих стадий схемы предполагает разборку по схеме ретрореакции Михаэля, которая будет рассмотрена ниже). Однако также очевидно, что, хотя синтез 74 в соответствии с показанной схемой разборки выглядит простым, эта простота на самом деле обманчива, ибо суть задачи в данном случае состояла в обеспечении полной стереоселективности образования каждой из вновь образующихся связей С—С. Поэтому в действительности потребовалось выполнить огромный объем исследований по разработке путей стереоспецифического проведения конденсаций альдольного типа и дизайну подходящих субстатов, что и позволило в конце концов эффективно выполнить синтез 74, почти не отклоняясь от принципиальной схемы, предложенной на основании достаточно очевидного регросинтетического анализа [11].
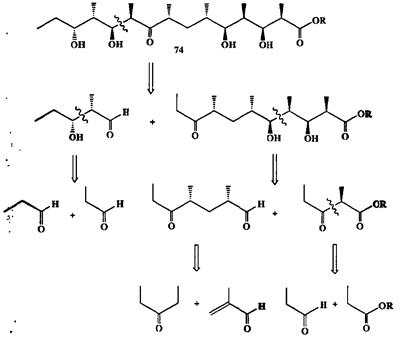 Схема 2.27
Схема 2.27
|
Конденсация двух карбонильных производных, так же как и реакция Гриньяра, относятся к одному фундаментальному классу органических реакций (нуклеофильнос присоединение), протекающих путем атаки карбаниона R" или его синтетического эквивалента по углеродному атому карбонильной группы. Вальдольной конденсации или реакции Гриньяра карбонильная группа выступает в роли эквивалента карбокатиона ^ С+-ОН, и в превращениях этого типа конечным результатом является образование одинарной связи С-С фрагмента R-C(OH). Существует еще ряд реакций, относящихся к тому же общему классу, однако в них карбонильная группа участвует как единое целое — как электрофил, эквивалентный бидентатному иону ^ С2+. Одна из важнейших конденсаций этого типа (реакция Виттига) присоединение илидов фосфора 75 по карбонильной группе альдегидов или кетонов [12а,B]. Начальная стадия этой реакции — атака карбанионного центра илида по атому углерода карбонильной группы — приводит к биполярному иону, бетаину 76 (схема 2.28). Структура этого диполя благоприятствует возможности взаимодействия противоположных зарядов на кислороде и фосфоре и возникновению ковалентной связи между ними, что в конце концов приводит к элиминированию трифе-нилфосфиноксида с одновременным образованием двойной связи между углеродными атомами участвующих реагентов. Поскольку требуемые илиды легко могут быть получены из соответствующих алкилгалогенидов, таких, как 77, через промежуточное образование фосфониевой соли 78, то суммарный синтетический результат применения метода Виттига может быть описан как сборка двойной связи из алкилгалогенида и карбонильного соединения.
Конечно, в принципе тот же результат мог бы быть достигнут последовательностью двух стадий, а именно: присоединением по Гриньяру и дегидратацией получающегося карбинола, однако последняя реакция, как правило, протекает с образованием смесей изомерных алкенов, что совершенно исключено самой природой реакции Виттига. На схеме 2.28 показан типичный пример получения метиленциклогексана (79) из циклогексанона, что практически нереально сделать с помощью показанной последовательности реакция Гринмра/дегидратация, но легко достижимо по реакции Виттига с использованием метиленфосфорана (80).
Алкилидснфосфораны типа 80 довольно нестабильны и поэтому их приходится генерировать in situ. Стабильность этих частиц резко возрастает при наличии дополнительных электроноакцепторных заместителей у карбанионного центра, и производные типа 81 уже могут быть выделены в свободном состоянии, и многие из них доступны в виде продажных реагентов. Реакция Виттига с помощью таких реагентов служит удобным путем синтеза а,р-непредельных карбонильных соединений, таких, как 82 (схема 2.28). Дополнительным преимуществом этого метода является возможность обеспечения строгого контроля стереохимии образующейся двойной связи.
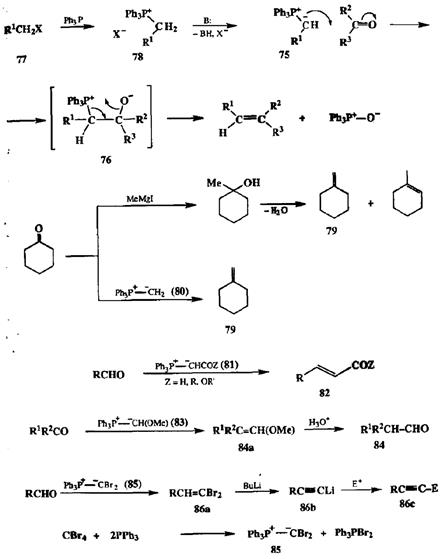 Схема 2.28
Схема 2.28
|
Открытие реакции Виттига в 1953 г. [ 12Ь](принесшее ее автору Нобелевскую премию) и последующие разработки ряда модификаций этого метода [12с] существенно расширили арсенал органического синтеза, снабдив синтетиков чрезвычайно мощным инструментом для синтеза олефинов со строго определенным расположением и стереохимией образующейся двойной связи. Благодаря этому стадия разборки двойной связи с выходом к паре предшественников — алкилгалогенид и карбонильное производное (ретрореакция Виттига) — является в настоящее время одним из наиболее надежных приемов ретросинтетического анализа самых разнообразных соединений.
Уместно также отметить, что универсальность реакции Виттига открывает ряд совершенно новых возможностей синтетического применения карбонильных производных. Типичной иллюстрацией могут служить два приведенных ниже примера.
Так, использование метоксиметиленфосфорана (83) (легко получаемого из хлорметилового эфира) в качестве илидной компоненты стало сейчас стандартным путем трансформации альдегидов или кетонов в гомологичные альдегиды типа 84 через стадию образования промежуточного винилового эфира 84а [12а] (схема 2.28).
Другой пример показывает возможность применения метода Виттига в синтезе ацетиленовых призводных, для чего требуется использовать в качестве реагента Виттига дибромметиленфосфоран (85). Оказалось, что этот реагент легко может быть получен взаимодействием трифенилфосфина с тетрабромметаном [12d]. Непосредственным результатом реакции соединения 85 с альдегидами является образование 1,1-дибромалкенов 8ба. Последние под действием бутиллития могут быть превращены в ацетилениды 86Ь, которые в свою очередь по стандартной реакции с электрофилами образуют ацетиленовые производные типа 86с {12е]. Показанная последовательность используется сейчас как удобный путь получения различных ацетиленовых соединений из легко доступных альдегидов.
Дата добавления: 2015-04-05; просмотров: 1882;