Серповидно-клеточная анемия (СКА)
Тип наследования аутосомно-рецессивный, дефект бета-глобиновых цепей, в которых в 6 положении гидрофильный глютамин заменен гидрофобным валином, образуется HbS. Восстановленная форма HbS мало растворима. При гипоксемии и снижении скорости кровотока HbS полимеризуется в длинные нерастворимые нити, растягивающие эритроциты в форме серпа. Если содержание HbS больше 45% (гомозиготное состояние), образуются эритроциты необратимо серповидной формы, склонные к агрегации, что повышает вязкость крови, вызывает закупорку сосудов (вазоокклюзию), нарушение микроциркуляции и боль.

В этом случае точечная мутация обусловливает нарушение структуры гемоглобина, гемолиз, анемию, а также закупорку сосудов и нарушение кровообращения.

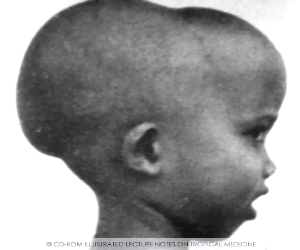
Больные СКА имеют типичный вид: удлиненный нижний сегмент тела, выступающий лоб, «башенный» череп, гепато-спленомегалия. Самый характерный криз для этого заболевания - вазоокклюзионный, проявляющийся резкой болью. Окклюзия сосудов может развиваться в разных органах, поэтому клинические симптомы чрезвычайно разнообразны. В мазках крови можно обнаружить серповидные клетки.

Примерно одна треть обитателей тропических и субтропических регионов Африки (т. наз. «малярийного пояса») являются носителями признака серповидноклеточности. Они не страдают от гемолитической анемии, в то же время более устойчивы по отношению к малярии.
Талассемии - замедление или отсутствие синтеза одной из цепей глобина:
α-талассемия, ß-талассемия.
При талассемиях мутации располагаются не в структурных генах, а в генах-регуляторах, поэтому структурных нарушений нет, а результатом мутаций служит замедление или отсутствие синтеза одной из глобиновых цепей и замена ее синтезом другой цепи. Талассемия встречается в странах Средиземноморья, в Китае, Индии, в Европе, у жителей Закавказья и Средней Азии. Самая высокая заболеваемость – на Мальдивах, где носители признака составляют 18%. Гетерозиготная бета-талассемия наблюдается у 7— 10% населения в низменных районах Азербайджана.
Альфа-талассемия – полное или частичное прекращение синтеза α-цепей. Компенсаторно синтезируются: а) в пренатальный период γ-цепи - образуется тетрамер γ (Hb Барт); б) в постнатальный – тетрамер ß (HbH).
Синтез α-цепей кодируют 4 гена, поэтому степень нарушения их синтеза меньше, чем при ß-талассемии; выраженный дисбаланс развивается только тогда, когда поражены все 4 гена. Агрегаты из ß-цепей более растворимы, чем агрегаты из α -цепей, поэтому гемолиз при α-талассемии выражен слабее, чем при ß-талассемии, а эритропоэз более эффективен.
Бета-талассемия обусловлена снижением скорости синтеза ß-цепей гемоглобина (ß+-талассемия) или отсутствием их синтеза (ß0-талассемия). Неповрежденные α-цепи избыточно накапливаются в клетках, что ведет к повреждению мембраны и разрушению эритроидных клеток в костном мозге (неэффективный эритропоэз) и эритроцитов в крови. Деструкция эритроидных клеток способствует гиперплазии костного мозга, что отражается на структуре скелета, ведет к повышенному всасыванию железа и перегрузке организма железом.
Тяжелая гомозиготная форма ß-талассемии - болезнь Кули, или большая талассемия. Кроме того, выделяют промежуточную, малую и минимальную талассемию.
 Для тяжелых форм талассемий характерна:
Для тяжелых форм талассемий характерна:
значительная спленомегалия,

желтушность, хронические язвы на нижних конечностях, башенный череп,
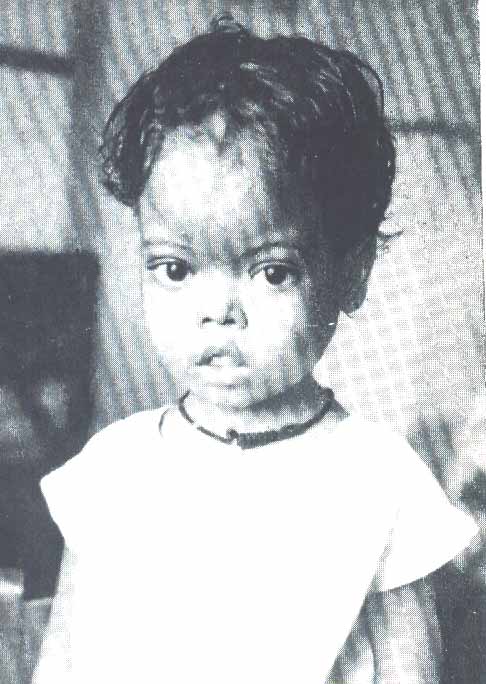
уплощенная переносица. Скулы выступают, глазные щели сужены, нарушены прикус и расположение зубов.

 Картина крови: мишеневидные эритроциты, анизоцитоз, пойкилоцитоз
Картина крови: мишеневидные эритроциты, анизоцитоз, пойкилоцитоз
.

Сканирующая электронограмма. Стрелками показаны два кодоцита («хвостатые клетки») – это другое название мишеневидных клеток.
Дата добавления: 2015-05-08; просмотров: 1577;