Наследственные заболевания хрусталика
Причиной наследственных заболеваний могут быть генные, хромосомные и геномные мутации. Патологии хрусталика уделено большое место в каталогах наследственных заболеваний. Среди последних наиболее часто встречаются катаракты. Значительно реже – вывихи и подвывихи хрусталика и др.
Врожденная катаракта чаще наследуется по аутосомно-доминантному типу, однако возможна и аутосомно-рецессивная передача, которая чаще встречается при кровном родстве родителей. При рецессивном, сцепленном с полом, наследовании болеют мужчины, а переносчиками болезни являются женщины. Реже встречаются сцепленное с полом доминантное наследование, когда заболевание проявляется только у дочерей.
Значительную группу составляют врожденные или рано приобретенные заболевания хрусталика при наследственных нарушениях обмена веществ.
Заболевания хрусталика при наследственных нарушениях обмена веществ. Среди наследственных заболеваний значительную группу составляют болезни, являющиеся следствием нарушения обменных процессов. При этих заболеваниях наряду с поражением различных органов и систем часто наблюдается патология органа зрения, в частности хрусталика.
Катаракта при наследственных нарушениях углеводного обмена. Катаракта при галактоземии. Катаракта является ранним признаком галактоземии – наследственного нарушения углеводного обмена. Заболевание наследуется по аутосомно-рецессивному типу. При галактоземии нарушен синтез фермента галактозо-1-фосфат-уридинтрансферазы. Отсутствие этого фермента препятствует превращению галактозы, входящей в состав молочного сахара (лактозы), содержащегося в молоке, в глюкозу. В результате этого в крови и тканях, в частности в хрусталике, накапливается галактоза и галактлзо-1-фосфат, оказывающие токсическое действие на ткани.
Основными признаками галактоземии, возникающими в первые дни или недели после рождения, являются потеря аппетита, рвота, понос, желтуха, гепатоспленомегалия, истощение, цирроз печени. Наблюдается галактозурия, альбуминурия, гипераминоацидурия, отсутствие галактозо-1-фосфат-уридинтрансферазы в эритроцитах. При тяжелых формах заболевания может быть летальный исход в первые месяцы жизни.
Примерно у 2/ 3 больных развивается двусторонняя катаракта, которую обнаруживают в первые недели жизни, как правило, до 3 – 6 мес. Катаракта вначале ядерная или слоистая, быстро прогрессирует, превращаясь в полную. Катаракта может возникнуть и в юношеском возрасте при врожденном отсутствии галактокиназы в эритроцитах.
Патологические процессы при галактоземии можно предотвратить путем перевода ребенка на диету, исключающую молоко и молочные продукты. При своевременном лечении в начальном периоде развития катаракты (у детей до 3 мес) хрусталик может просветляться, после 6 мес. жизни образование катаракты и развитие других проявлений болезни предотвратить не удается.
Катаракта при гипогликемии. Помутнение хрусталика может развиваться у детей раннего возраста при гипогликемии. Считают, что недостаток концентрации глюкозы как в крови, так и в спинномозговой жидкости может быть причиной поражения головного мозга и хрусталиков. В то же время не исключают, что поражение этих структур является следствием пренатальной или постнатальной аноксии и гипогликемии. Проведение своевременного лечения сможет предотвратить развитие катаракты у тех детей, у которых при рождении хрусталики нормальные.
Катаракта при диабете. Диабет в большинстве случев наследуется по аутосомно-рецессивному типу. В основе заболевания лежат врожденная и наследственная неполноценность островкового аппарата поджелудочной железы и провоцирующие факторы внешней среды. Наиболее часто дети заболевают в возрасте от 6 до 13 лет, однако заболевание может быть и у грудных детей. Катаракта развивается у 1 – 4% больных диабетом и у детей наблюдается чаще, чем у взрослых. Помутнение хрусталика почти всегда бывает двусторонним и быстро прогрессирует. Развитие катаракты у детей, страдающих диабетом, свидетельствует о тяжелом течении заболевания.
Катаракта при маннозидозе. Помутнение хрусталика является одним из проявлений маннозидоза – врожденного нарушения метаболизма, связанного с дефицитом активной кислой маннозидазы А и В. При этом наблюдается лизосомальная аккумуляция богатых маннозой гликопептидов, гликопротеидов и олигосахаров в тканях и средах организма, Заболевание передается по аутосомно-рецессивному типу. При маннозидозе могут наблюдаться катаракта, поверхностные помутнения роговицы, бледный или сероватый диск зрительного нерва и нечеткость его границ, сходящееся косоглазие. Начальные изменения хрусталика заключаются в спицеобразных помутнениях под задней капсулой или в точечных помутнениях линзы. Одновременно наблюдается задержка психомоторного развития, лицевые нарушения, множественные дизостозы, гепатоспленомегалия, понижение слуха, повышенная восприимчивость к инфекциям.
Катаракта при нарушениях кальциевого обмена (тетаническая). Нарушения кальциевого обмена наблюдаются при тетании, спазмофилии, рахите, почечной недостаточности. Преимущественной формой катаракты является слоистая (зонулярная). Помутнение хрусталика всегда двустороннее. Лечение (внутривенные инъекции 10% раствора хлорида кальция, препараты щитовидной железы), начатое в ранней стадии заболевания, может задержать развитие катаракты.
Поражение хрусталика при наследственных нарушениях метаболизма соединительной ткани и аномалиях костной системы.
Синдром Марфана – наследственное заболевание, обусловленное нарушением метаболизма соединительной ткани. Передается по аутосомно-доминантному типу. Биохимическими исследованиями установлено, что при синдроме Марфана наблюдается усиленная экскреция с мочой оксипролина и кислых гликозаминогликанов (КГАГ).
Синдром Марфана представляет собой сочетание изменений глаз с поражением сердечно-сосудистой, костно-мышечной и других систем организма с расстройствами деятельности желез внутренней секреции, а также психики. Поражение сердечно-сосудистой системы заключается в пороках сердца, аневризмах аорты, тромбозах вен и др. Изменениями костно-мышечной системы является арахнодактилия ( длинные пальцы рук и ног), долихофацилия, ломкость костей , частые вывихи вследствие слабости связочного аппарата. Наблюдается непомерно высокий рост, тонкие и непропорционально длинные кости конечностей, контрактуры суставов, сколиозы, кифозы, воронкообразная грудь, крыловидные лопатки, недоразвитие подкожного жирового слоя, мышц, половых органов, варикозные узлы, опущение органов, удлинение кишечника и др. Наиболее часто встречаются поражения глаз, деформация скелета и сердечно-сосудистые нарушения. Наряду с типичными формами заболевания, могут встречаться стертые, характеризующиеся наличием одного –двух симптомов. В связи с поражением ряда систем больные нередко умирают в раннем детском возрасте, но часто доживают до зрелого и даже пожилого возраста (рис.1).
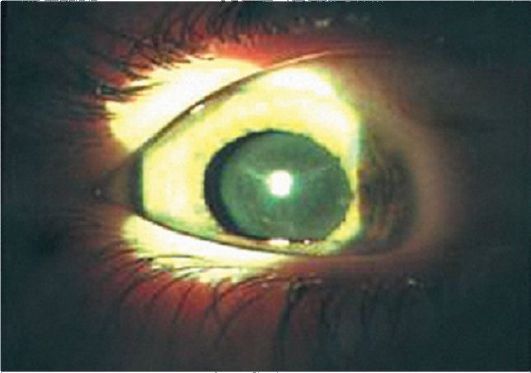
Рис. 1. Синдром Марфана. Сублюксация полупрозрачного хрусталика кверху. (Собст. фото)
Поражение органа зрения при синдроме Марфана встречается в 50 – 100% случаев, нередко являясь одним из ранних признаков заболевания, предшествующих общим клиническим симптомам. Могут наблюдаться эктопия хрусталика, сферофакия, катаракта, кератоконус, фибриллярная и зернистая деструкция стекловидного тела, дегенерация и отслойка сетчатки, миопия, глаукома, косоглазие, нистагм и др.
Ранними признаками синдрома Марфана являются эмбриотоксон, гипоплазия радужки, особенно ее пигментной каймы.
Эктопия хрусталика является одним из наиболее частых проявлений поражения органа зрения при этом заболевании. Встречается почти в 80% случаев. Характерным является преимущественное смещение хрусталиков чаще кверху-кнаружи, кверху-кнутри, реже – книзу, всегда двустороннее. Эктопия хрусталика постепенно прогрессирует, вследствие чего увеличивается степень смещения его вплоть до вывиха в переднюю камеру.
Гомоцистинурия – наследственное заболевание, в основе которого лежит нарушение обмена серосодержащих аминокислот, связанное с недостаточностью фермента цистатаионсинтетазы. В крови обнаруживается большое количество гомоцистина. Повышена экскреция с мочой КГАГ и оксипролина. Заболевание наследуется по аутосомно-рецессивному типу и характеризуется умственной отсталостью и судорогами.
Глазными проявлениями являются эктопия хрусталиков книзу, связанная с деструкцией ресничных поясков, сферофакия, врожденная катаракта, дегенерация сетчатки, атрофия зрительного нерва, миопия высокой степени и др.
Синдром Маркезани наследуется по доминантному и аутосомно-рецессивному типу. По многим признакам противоположен синдрому Марфана. Характерными проявлениями заболевания являются маленький рост, укороченные туловище, шея и конечности, брахицефалия, ограничение подвижности суставов, расстройства сердечно-сосудистой системы. Со стороны органа зрения наблюдаются эктопия хрусталиков книзу, сферофакия, микрофакия, прогрессирующая близорукость высокой степени, отслойка сетчатки, вторичная глаукома.
Синдром Элерса-Данлоса - наследственное заболевание, которое связывают с поражением коллагеновой и эластичной ткани, Наследуется по доминантному и аутосомно-рецессивному типу. Характеризуется повышенной ранимостью кожи, ломкостью поверхностных сосудов, рецидивирующими гематомами, образованием кожных рубцов, чрезмерной подвижностью суставов, вывихами тазобедренных суставов, аномалиями внутренних органов (грыжи, дивертикулы желудочно-кишечного тракта), сердечно-сосудистыми нарушениями, Глазными проявлениями являются блефарохалязис, отек и атрофия кожи век, эпикантус, склеры голубого цвета, дистрофия роговицы, кератоконус, микрокорнеа, подвывихи хрусталика, глаукома, отслойка сетчатки и др.
Врожденная хондродистрофия – наследственное заболевание, характеризующееся нарушением и замедлением эпифизарного роста длинных костей при нормальном периостальном росте, что ведет к укорочению и утолщению костей. Характерно длинное туловище с укороченными деформированными конечностями, брахицефальная форма черепа, короткие утолщенные пальцы, деформация и тугопдвижность суставов. Возможны другие аномалии (врожденный порок сердца, полидактилия, заячья губа и др.). Одним из самых постоянных симптомов является врожденная катаракта. Возможны другие изменения органа зрения (атрофия зрительного нерва).
Синдром Апера (акро-цефало-синдактилия, синдром первой жаберной дуги) – наследственное заболевание, передающееся, как правило, по аутосомно-доминантному типу. Основными симптомами заболевания являются деформация черепа (башенный), широкое основание носа, синдактилия, полидактилия, гипоплазия верхней челюсти, порок сердца, умственная отсталость. Изменениями органа зрения могут быть уплощение орбит, экзофтальм, расходящееся косоглазие, нистагм, колобома сосудистой оболочки, поражение сетчатки и зрительного нерва (застойные диски, атрофия). Нередко наблюдается катаракта.
Синдром Конради-Хюнерманна (врожденный точечный кальциноз эпифизов) – наследственное заболевание, передающееся по аутосомно-рецессивному типу, основными симптомами которого являются непропорциональный нанизм, аномалии пальцев, сколиоз, поражение связочного аппарата суставов. При рентгенологическом исследовании выявляется точечное обызвествление эпифизов. Почти у половины больных наблюдается катаракта (полная или реже центральная), которая, как правило. Обнаруживается сразу после рождения. В отдельных случаях помутнение хрусталика может развиться на первом году жизни.
Катаракта при наследственных поражениях кожи. Развитие врожденной катаракты при некоторых поражениях кожи связывают с тем, что хрусталик является производным эктодермы.
Синдром Ротмунда характризуется дерматозом, катарактой и гипогенитализмом, встречается чаще у девочек, развивается в возрасте от 3 до 6 мес, передается по аутосомно-рецессивному типу. Наблюдается поражение желез внутренней секреции (гипофиза, щитовидной железы, половых желез).
Врожденный ихтиоз кожи с гиперкератозом нередко сопровождается поражением органа зрения (врожденная катаракта, микрофтальм, ороговение эпителия роговицы, изменения век).
Синдром Шефера характеризуется ладонно-подошвенным и рассеянным фолликулярным кератозом кожи, очагами облысения, микроцефалией, карликовым ростом, олигофренией, гипогенитализмом, врожденной катарактой. Передается по аутосомно-доминантному типу.
Инфантильный пигментный дерматоз (болезнь Блоха-Сульцберга) – наследственное заболевание с аутосомно-доминантной передачей. Развивается, как правило, в течение первой недели жизни, реже на первом году, встречается чаще у девочек. Характеризуется пигментацией кожи, облысением, аномалиями зубов, врожденной катарактой, наряду с которой могут встречаться другие поражения глаз (косоглазие, нистагм, атрофия зрительных нервов).
Конституциональная шаровидно-клеточная гемолитическая анемия – заболевание с аутосомно-доминантной передачей. Основными симптомами являются гемолитическая желтуха, спленомегалия, костные аномалии, нарушения пигментации кожных покровов, аномалии зубов, отосклероз, полидактилия, поражение органа зрения (врожденная катаракта, микрофтальм, гетерохромия, эктопия зрачка, помутнение роговицы, цветослепота).
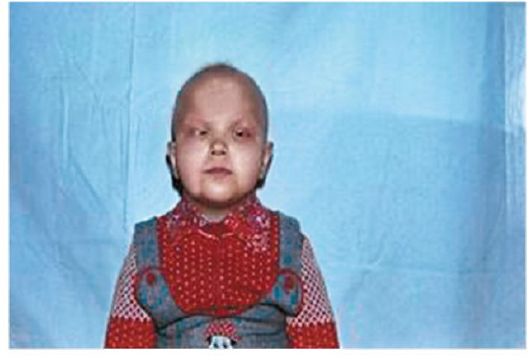
Синдром Сабуро – двустороннее помутнение хрусталиков, развивающееся на 2 – 5 году жизни в сочетании с гипотрихозом, переходящим в тотальную алопецию. Наследуется по доминантному и рецессивному типу (рис.2).
Рис. 2. Ребенок с синдромом Сабуро: врожденная атипичная катаракта, микрокорнеа, кератоз, гипотрихоз (отсутствие бровей, ресниц, волос на голове), брахидактилия, синдактилия пальцев ног и др. (Собст. фото)
Наследственные хромосомные заболевания с поражением хрусталика. Врожденная катаракта может быть одним из проявлением ряда наследственных заболеваний или синдромов, в основе которых лежит нарушение хромосомного набора. Могут наблюдаться изменения структуры, положения или числа хромосом. Хромосомные заболевания чаще отмечают при рождении детей от немолодых матерей.
Болезнь Дауна. Заболевание обусловлено трисомией хромосомы 21, в редких случаях может быть транслокация хромосомы 21 на другую хромосому. Представляет тяжелую аномалию соматического и умственного развития. Характерными признаками являются идиотия или имбецильность, микро- и брахицефалия, широкая запавшая переносица, «географический язык», недоразвитие верхней челюсти, прогнатизм, малый рост или акромегалия, гипотиреоз, ихтиоз, кератоз, алопеция, гипогенитализм, синдактилия, полидактилия, врожденный порок сердца и другие аномалии. Около половины больных умирают преимущественно на первом году жизни вследствие малой устойчивости к инфекциям. В то же время некоторые больные доживают до глубокой старости.
Со стороны органа зрения наблюдаются эпикантус, косая глазная щель. Нистагм, косоглазие, помутнение роговицы. Катаракта ( полная или частичная) встречается у 15 – 50% больных.
Синдром Маринеску – Съегрена – наследственное заболевание с аутосомно-рецессивной передачей. Характеризуется резкой задержкой физического и психического развития, врожденной катарактой с косоглазием, нистагмом и другими изменениями органа зрения, врожденной мозжечково-спинальной атаксией, карликовым ростом.
Синдром Шершевского-Тернера (синдром птеригиума шеи). Хромосомное заболевание, при котором отсутствует Y-хромосома при наличии лишь одной Х-хромосомы. Наблюдаются крыловидные складки на шее, нанизм, умственная отсталость, дискрания, синдактилия, аномалии ушных раковин, лимфоангиоэктатические отеки, врожденная катаракта, микрофтальм, парез отводящего нерва, косоглазие, малые размеры диска зрительного нерва и др.
Синдром Лоу (окулоцереброренальный). Тип наследования аутосомно-рецессивный, сцепленный с полом. Заболевание наблюдается у мальчиков, Характеризуется расстройствами выделительной функции почек, крипторхизмом, задержкой окостенения скелета, умственной отсталостью, альбуминурией, аминоацидурией, вторичной глюкозурией.
Глазными проявлениями являются двусторонняя врожденная катаракта, микрофакия. Дистрофия роговицы, повышение внутриглазного давления, склеры голубого цвета, качательный нистаг, расходящееся косоглазие.
Болезнь Норри. Заболевание вызвано сцепленным с полом рецессивным геном. Страдают только лица мужского пола. Болезнь проявляется в первые месяцы жизни, поражаются оба глаза. За хрусталиком образуются белого цвета васкуляризированные массы (псевдоглиома), появляются катаракта, синехии, поражения сетчатки, позже развивается атрофия глазного яблока. Наблюдаются умственная отсталость и прогрессирующая глухота.
Редкие синдромы с наличием катаракты.
Синдром Аксенфельда – сочетание помутнений хрусталика, колобомы радужки, аниридии с повышением ВГД.
Синдром Альпорта (нефропатия с глухотой, семейная) – отоокулоренальный синдром, наследственный геморрагический нефрит. Болеют исключительно мальчики. Наблюдаются глухота, поражение почек и глаз (катаракта, сферофакия, пигментная дегенерация).
Синдром Кокейна – карликовый рост, микроцефалия, атрофия подкожной клетчатки на лице. Глазными проявлениями являются катаракта, пигментная дегенерация сетчатки, атрофия зрительного нерва. Наследуется по рецессивному типу.
Синдром Ригера – сочетание гипоплазии мезодермального листка радужки, деформации зрачка с аномалиями угла передней камеры и вторичной глаукомой. Могут встречасться эктопия хрусталика, врожденная катаракта, колобомы сосудистой оболочки, дермоидные кисты у лимба.
Синдром Сименса – сочетание пигментного дерматита и врожденной катаракты при низком росте. Доминантный тип наследования, связанный с полом.
Синдром Халлерманна – врожденная катаракта в сочетании с брахицефалией, птичьим носом, микрофтальмом, микрофтальмией, гипоплазией нижней челюсти. Наследуется по рецессивному типу.
Одним из редких синдромов является Синдром Мартина-Олбрайта (рис.3).
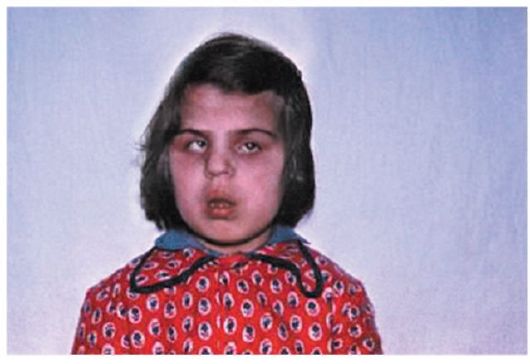
Рис.3. Синдром Мартина-Олбрайта-псевдогипопаратиреоз. Прогрессирующее помутнение хрусталиков в 4-6-летнем возрасте, пигментная дегенерация сетчатки. В крови - гипокальциемия и гипофосфатемия. Олигофрения, судороги, отставание в росте. (Собст. фото)
Дата добавления: 2016-03-20; просмотров: 1810;