Этапы клинического исследования ЛС
И производитель, и общество заинтересованы в том, чтобы в ходе исследований, предшествующих регистрации, была получена как можно более точная и полная информация о клинической фармакологии, терапевтической эффективности и безопасности нового ЛС. Подготовка регистрационного досье невозможна без ответа на эти вопросы. В связи с этим регистрации нового лекарственного средства предшествуют несколько десятков различных исследований, причем с каждым годом увеличивается как число исследований (рис. 3.1), так и число участников исследования (рис. 3.2). Общий цикл исследований нового ЛС обычно превышает 10 лет (рис. 3.3). В связи с этим разработка новых ЛС становится уделом только крупных фармацевтических компаний, а общая стоимость исследовательского проекта превышает 350 млн долларов.
| Рис. 3.1. Среднее число исследований, предшествующих регистрации новых препаратов (Food & Drug Law J, Vol. 52, 1997). |
Первые, доклинические исследования начинаются вскоре после синтеза новой, потенциально эффективной молекулы. Их суть заключается в проверке гипотезы о предполагаемом фармакологическом действии нового соединения. Параллельно изучают токсичность соединения, онкогенное и тератогенное действие. Все эти исследования выполняют на лабораторных животных, а их общая продолжительность составляет 5-6 лет. В результате такой работы из 5000—10 000 новых соединений отбирается около 250. Применение мето-


|
| Рис. 3.2. Среднее число пациентов, включенных в клинические исследования новых препаратов (Food & Drug Law J, Vol. 52, 1997). |
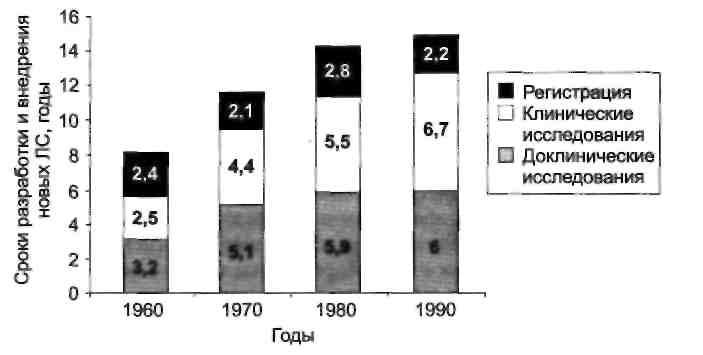
Рис. 3.3. Время, необходимое для разработки и внедрения нового ЛС (Tufts Center for the Study of Drug Development, 1998).
дов генной инженерии способно несколько сократить этот исследовательский период.
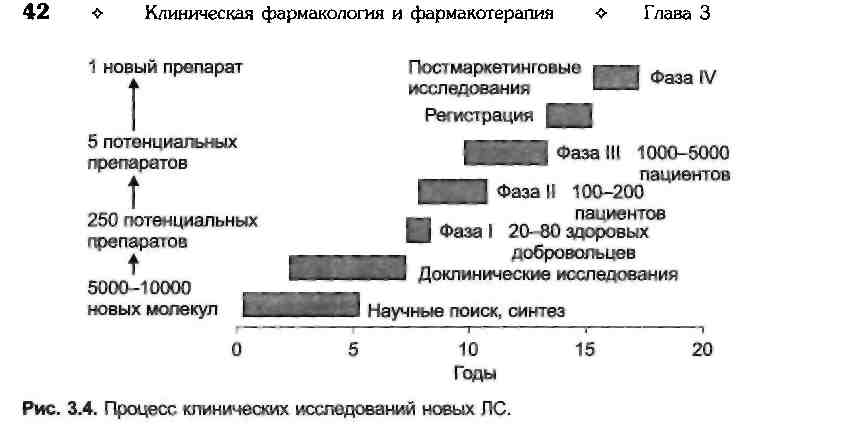
Собственно клинические исследования условно разделяют на 4 периода или фазы (рис. 3.4).
I фаза клинических исследований,как правило, проводится на 28-30 здоровых добровольцах. Цель заключается в получении сведений о фармакокинети-ке и фармакодинамике потенциального ЛС, уточнении режима дозирования и получении данных по безопасности препарата. Изучение терапевтического действия препарата в этой фазе необязательно, так как у здоровых добровольцев ряд клинически важных свойств нового ЛС не наблюдается.
Исследования I фазы начинают с изучения безопасности и фармакокине-тики однократной дозы, при выборе которой используют данные, полученные на биологических моделях. В дальнейшем изучают фармакокинетику препарата при многократном назначении, экскрецию и метаболизм нового ЛС (порядок кинетических процессов; см. главу 5), его распределение в жидкостях и
тканях организма, фармакодинамику. Обычно все эти исследования проводят для различных доз, лекарственных форм и путей введения; в ходе I фазы исследований можно также оценивать влияние на фармакокинетику и фармакодинамику нового препарата других ЛС, функционального состояния организма, приема пиши ит.д.
Важной целью I фазы клинических испытаний служит выявление потенциальной токсичности и нежелательных лекарственных реакций, но эти исследования непродолжительны и проводятся у ограниченного числа участников, поэтому в ходе этой фазы удается выявить только наиболее частые и выраженные нежелательные явления, связанные с применением нового ЛС.
В ряде случаев (онкологические препараты, препараты для терапии ВИЧ-инфекции и т.п.) исследования I фазы можно проводить у больных. Это позволяет ускорить создание нового препарата и не подвергать добровольцев необоснованному риску, хотя такой подход является скорее исключением.
Исследования I фазы позволяют:
оценить переносимость и безопасность нового препарата; в ряде случаев получить представления о его фармакокинетике (у здоровых людей, что, естественно имеет ограниченное значение);
• определить основные фармакокинетические константы (С^, Т, „ О и
т.д.; см. главу 5);
сравнить фармакокинетику нового препарата при использовании различных лекарственных форм, путей и способов назначения.
Исследования II фазы- первые исследования у больных. Объем этих исследований значительно больше, чем при в I фазе, 100-200 (до 500) пациентов. Во II фазе уточняют эффективность и безопасность нового препарата, а также диапазон доз для лечения больных. Эти исследования дают информацию в основном о фармакодинамике нового ЛС. Обязательными условиями проведения исследований II фазы являются сравнительный дизайн и включение контрольной группы (что нехарактерно для исследований I фазы).
Исследования II фазы подразделяют на:
• фазу Па - небольшие по числу пациентов пробные (pilot trial) иссле
дования. Проведение этих исследований позволяет продемонстрировать по-
Клинические исследования и регистрация новых /1С ♦ 43
тенциальную эффективность нового препарата для того, чтобы решить вопрос о целесообразности его дальнейшей разработки и планирования дальнейших более дорогостоящих исследований;
• фазу IIб — базовые клинические исследования (pivotal trial), в ходе
которых доказывается эффективность нового препарата для лечения или про
филактики какого-либо заболевания.
Исследования III фазы планируются у большого числа больных (до 10 000 и более), а условия их проведения максимально приближены к обычным условиям лечения больных с определенным заболеванием. Исследования этой фазы (как правило, это несколько параллельно или последовательно проводимых исследований) крупные (полномасштабные), рандомизированные и сравнительные. Предметом изучения становится не только фармакодинамика нового ЛС, но и его клиническая эффективность1.
В III фазе исследований препарат сравнивают по эффективности и безопасности с плацебо (плацебо-контролируемое исследование) или/и с другим маркерным препаратом, т.е. с ЛС, обычно используемым в данной клинической ситуации и обладающим хорошо известными лечебными свойствами.
Подача компанией-разработчиком заявки на регистрацию ЛС не означает завершения исследований. Исследования III фазы, выполненные до подачи заявки, называют исследованиями Ша фазы, а после подачи заявки — III6 фазы. Последние проводятся для получения более полной информации о клинической и фармакоэкономической эффективности ЛС. Такие исследования могут расширить показания к назначению нового ЛС. Инициатором дополнительных исследований могут стать и государственные органы, отвечающие за процесс регистрации, если результаты предшествовавших исследований не позволяют однозначно высказаться о свойствах и безопасности нового ЛС.
К концу II фазы из 250 перспективных соединений остаются только 4-5, дальнейшая разработка которых представляет интерес. Исследования Ш фазы проводят у большего числа больных, что позволяет точнее оценить терапевтическую эффективность нового препарата в сравнении с существующими. Одновременно собирают дополнительный материал по нежелательным лекарственным реакциям и определяют область применения нового препарата. В итоге регистрации достигает только 1 препарат из 5000—10 000 потенциально активных соединений.
Результаты исследований III фазы становятся определяющими при принятии решения о регистрации нового ЛС. Такое решение может быть принято, если препарат:
• более эффективен, чем уже известные ЛС аналогичного действия;
• обладает эффектами, которые не свойственны уже существующим препаратам;
• имеет более выгодную лекарственную форму;
' Например, цель исследования нового гипотензивного препарата в I—II фазах — доказать его способность снижать уровень АД, а при исследовании III фазы целью является изучение влияния ЛС на артериальную гипертензию. В последнем случае наряду со снижением АД появляются другие точки оценки эффекта, например снижение смертности от сердечно-сосудистых заболеваний, профилактика осложнений артериальной гипертензии, повышение качества жизни больных и т.д.
44 о- Клиническая фармакология и фармакотерапия о- Глава 3
• более выгоден в фармакоэкономическом отношении (см. главу 12) или позволяет использовать более простые методы лечения;
• имеет преимущества при совместном применении с другими ЛС;
• имеет более простой способ применения.
Исследования IV фазы.Конкуренция с новыми препаратами заставляет продолжать исследования и после регистрации нового ЛС (постмаркетинговые исследования), чтобы подтвердить эффективность препарата и его место в фармакотерапии. Кроме того, исследования PV фазы позволяют ответить на некоторые вопросы, возникающие в ходе использования ЛС (оптимальная продолжительность лечения, преимущества и недостатки нового препарата в сравнении с другими, в том числе более новыми ЛС, особенности применения ЛС у пожилых, детей, отдаленные эффекты лечения, новые показания и тл.).
В ряде случаев исследования IV фазы проводят спустя много лет после регистрации ЛС. Примером подобных отсроченных более чем на 60 лет исследований являются исследования нитроглицерина и других нитратов'.
Клинические исследования всех фаз проводят в официально сертифицированных государственными органами контроля2 центрах (медицинские центры, больницы, поликлиники), имеющих соответствующее научно-диагностическое оснащение и возможность оказания квалифицированной медицинской помощи больным с нежелательными лекарственными реакциями.
Несмотря на существенные затраты и строгую оценку эффективности, лишь 1 из каждых 10 зарегистрированных новых препаратов занимает лидирующее положение на рынке ЛС (например, лосег, золцер, липитор, норваск, пульми-корт), принося производителю значительную прибыль. Другие 8 новых зарегистрированных ЛС приблизительно окупают расходы на свое создание, и еще
1 препарат из 10 приносит убытки своему производителю и/или снимается с
производства (грепафлоксацин, липобай).
Исследования биоэквивалентности.Большинство препаратов на фармацевтическом рынке являются воспроизведенными (генерическими) препаратами. Фармакологическое действие и клиническая эффективность ЛС, входящих в состав этих ЛС, как правило, достаточно хорошо изучены. Однако эффективность генерических препаратов может существенно различаться3.
Регистрация генерических препаратов может быть упрошенной (по времени и по объему исследований). Сделать строго обоснованное заключение о качестве этих средств позволяют исследования биоэквивалентности. В этих исследованиях генерический препарат сравнивают с оригинальным по биодоступности (т.е. сравнивают доли препарата, достигающие системного крово-1 Предложенные в клиническую практику около 1 0 0 лет назад, эти препараты в свое время не проходили процесс регистрации и клинических исследований, что и потребовало их разносторонних исследований спустя более 60 лет. Современная система регистрации новых препаратов появилась в 60-х годах XX в., поэтому около 30—40% применяемых сегодня ЛС не были убедительно исследованы. Их место в фармакотерапии может быть предметом дискуссии. В англоязычной литературе для этих препаратов применяется термин «лекарства-сироты», так как редко удается найти источники финансирования для исследований таких ЛС.
2 В нашей стране Минздравом РФ.
3 На эффективность генерических ЛС влияют различия в технологии производства, недостаточ
ный контроль качества продукции, свойства наполнителей (таблетированные формы) и носите
лей и другие факторы, трудно поддающиеся идентификации.
Клинические исследования и регистрация новых /1С -fr 45
тока, и скорость, с которой этот процесс происходит). Если два ЛС обладают одинаковой биодоступностью (см. главу 5), они биоэквивалентны. При этом предполагают, что биоэквивалентные препараты обладают одинаковой эффективностью и безопасностью1.
Биоэквивалентность изучают на небольшом (20-30) числе здоровых добровольцев, при этом используют стандартные для исследования фармакоки-нетики процедуры (построение фармакокинетической кривой, исследования величины AUC, Т , С ; см. главу 5).
Дата добавления: 2016-03-15; просмотров: 2655;