Кистозная почка у младенцев и детей
Клинические признаки. Младенческая форма заболевания проявляется при рождении ребенка в виде диффузно увеличенных почек, почечной недостаточности и недоразвития внутрипеченочных желчных протоков. При детской форме заболевания наблюдается расширение расположенных в корковом веществе собирательных протоков, которое протекает бессимптомно и сочетается с врожденным фиброзом печени и портальной гипертензией. Наследуются обе формы заболевания по аутосомно-рецессивному типу. Почечная недостаточность часто развивается в обоих случаях, но смерть в детском возрасте наступает в результате поражения печени.
Морфология. Младенческая форма заболевания характеризуется расширением дистальных отделов канальцев и собирательных протоков и превращением их в растянутые кисты, располагающиеся в радиальном направлении, как правило, в корковом веществе почки. Это приводит к тому, что размер почек увеличивается, они приобретают губчатое строение. При детской форме заболевания число кист меньше, собирательные протоки, расположенные в корковом веществе почек, поражаются в меньшей степени, и размеры почек не столь сильно увеличены. Небольшие внутрипеченочные желчные протоки беспорядочно расширены, а области вблизи воротной вены заполняют крупные взаимосвязанные полости, выстланные гиперплазированным эпителием. При этой форме заболевания имеет место скорее портальный фиброз, а не дилатация и пролиферация мелких желчных протоков, и в позднем детском возрасте, как правило, развивается портальная гипертензия.
Диагностика и лечение. Кистозные почки у младенцев могут иметь достаточно большие размеры для того, чтобы вызвать затрудненное родоразрешение. При рождении они не функционируют и служат причиной развития олигурической почечной недостаточности, расстройства дыхания, гипертензии и застойной сердечной недостаточности. При внутривенной урографии можно получить пятнистую нефрограмму с различной степенью задержки рентгеноконтрастного вещества в кистах, соответствующей расширенным собирательным протокам, расположенным в корковом и мозговом веществе почек. При выполнении ретроградной урографии видно, что почечные чашечки имеют закругленные концы, наблюдается тубулярный рефлюкс. При детской форме заболевания результаты внутривенной урографии могут вызвать предположение о губчатых изменениях мозгового вещества почки вследствие выраженного расширения канальцев в мозговом веществе. Часто развиваются почечная недостаточность и хроническое инфекционное заболевание.
Губчатая почка
Патология. При этом состоянии протоки Беллини, т. е. терминальные собирательные протоки, которые достигают концов сосочков и по которым моча поступает в почечную лоханку, расширены до размера кист и часто содержат камни, состоящие из оксалата кальция. Почки при этом асимметричны, и более тяжело пораженная почка имеет больший размер. Около верхушки каждого пораженного сосочка обнаруживают одну или несколько медуллярных кист. Камни образуются в расположенных в терминальных собирательных протоках кистах или проксимальнее их (рис. 228-1). Изменения паренхимы вторичны по отношению к внутрипочечной обструкции. Кисты выстланы кубическим эпителием, а иногда псевдомногослойным и многослойным плоским эпителием.
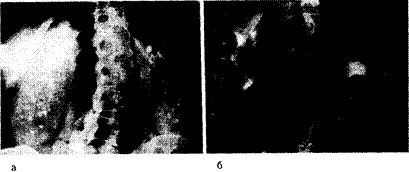
Рис. 228-1. Патологические изменения при губчатой почке. а — рентгенологическое изображение почки с губчатообразными изменениями мозгового вещества. На обзорной рентгенограмме брюшной полости обнаруживается множественная двусторонняя кальцификация; б — рентгеноконтрастное вещество накапливается в расширенных и пораженных кистами терминальных собирательных протоках и затемняет кальцификаты.
Диагностика и лечение. Губчатые изменения мозгового вещества почек отмечаются на 1 из 200 неотобранных внутривенных пиелограмм. Хотя большинство случаев заболевания спорадические, были описаны и случаи аутосомно-доминантного наследования. При губчатой почке различают две клинические картины в зависимости от возраста больного: одну в подростковом возрасте и другую в третьем или четвертом десятилетиях жизни. Образование камней, развитие инфекционного заболевания и гематурию отмечают в этих возрастных группах соответственно у 60, 35 и 30% больных. Часто встречаются сосочковый нефрокальциноз, обусловленнный наличием камней в кистах. Гиперкальцийурия развивается почти у половины из тех больных, у которых происходит образование камней, но она столь же распространена и при других видах болезни, связанной с образованием кальциевых камней (гл. 229). Гипертензия наблюдается не чаще, чем среди обычной популяции. Почечная недостаточность развивается редко, в основном у лиц с тяжелопротекающими почечнокаменной болезнью и/или инфекционным заболеванием почек.
Диагноз губчатой почки ставят на основании результатов внутривенной урографии. Выраженность обратного тока мочи из лоханки в канальцы различна: от простого покраснения сосочков до явного расширения канальцев у верхушек сосочков. Часто наблюдаются маленькие пирамидообразные кисты и нефрокальциноз, а сосочковые конкременты затемнены применяемыми при урографии рентгеноконтрастными средствами. Расширенные собирательные протоки с трудом заполняются во время ретроградной пиелографии, и поэтому рентгеноконтрастное вещество остается отделенным от сосочковых конкрементов в кистах.
Больные без явных симптомов заболевания не нуждаются в каком-либо лечении, им целесообразно лишь избегать гипогидратации организма, снижая тем самым риск образования камней. Следует попытаться выявить метаболическую этиологию образования камней и провести общепринятое лечение; лечение больных с инфекционным заболеванием и урологическими последствиями образования камней описано в гл. 229. Больные с губчатой почкой чрезвычайно восприимчивы к развитию инфекционных заболеваний, поэтому нужно свести к минимуму урологические инструментальные манипуляции.
Кисты мозгового вещества (нефронофтизный комплекс)
Этиология. Существуют некоторые виды наследственных кистозных поражений мозгового вещества почек, имеющих сходные морфологические признаки, но отличающиеся друг от друга по типу наследования. Рецессивно наследуемая форма этого поражения связана с развитием у больного почечной недостаточности до достижения им 20-летнего возраста, в то время как доминантно наследуемая форма поражения вызывает развитие почечной недостаточности только после достижения больным 20-летнего возраста. Заболевание почек, сопровождающееся дегенеративными изменениями сетчатки (почечная дисплазия сетчатки), наследуется по рецессивному типу, хотя почечная недостаточность развивается в этих случаях во взрослой жизни.
Патология. При обеих формах заболевания большая часть кист расположена в мозговом веществе и в кортикомедуллярной области и локализована в собирательных протоках и дистальных отделах извитых канальцев. Кисты выстланы слоем атрофичного эпителия, их диаметр может достигать нескольких миллиметров. Почки асимметрично сморщены и покрыты рубцами. Имеют место атрофия канальцев и перигломерулярный фиброз (атрофия выражена значительнее). В далеко зашедших случаях заболевания происходят склероз и гиалиноз клубочков, фиброз коркового вещества и клеточная инфильтрация интерстиция, и в этом случае гистологическую картину трудно отличить от таковой при хроническом интерстициальном нефрите.
Диагностика и лечение. Повреждение дистальных отделов нефрона приводит к нарушению концентрированной способности, а также процессов экскреции кислот и сохранения в организме натрия. Заболевание характеризуется полиурией, прогрессирующей почечной недостаточностью, низкорослостью, тяжелой анемией, гиперхлоремическим метаболическим ацидозом и снижением способности удерживать натрий в организме. У взрослых неспособность к сохранению натрия в организме может вызвать развитие синдрома, связанного с истощением запасов натрия, который напоминает признаки недостаточности надпочечников, но не поддается коррекции минералокортикоидами. В итоге развивается гипертензия. В начале заболевания моча без изменений, но впоследствии может развиваться протеинурия. При проведении внутривенной урографии обнаруживают почки малых размеров, покрытые рубцами, но без кальцификации. Чашечки деформированы многочисленными кистами, расположенными в кортикомедуллярной области.
В организм больного необходимо вводить большие количества натрия и воды, а также щелочи для коррекции ацидоза. Лечение при инфекционных осложнениях, анемии, гипертензии и других проявлениях терминальной стадии почечной недостаточности рассмотрено в гл. 220. При планировании семьи и отборе здорового донора-родственника для трансплантации почки может принести пользу генетическая консультация.
Синдром Барттера
Синдром Барттера включает в себя гипокалиемию, обусловленную утратой большого количества калия с мочой, повышение активности ренина в плазме крови и секреции альдостерона, нормальную величину артериального давления, сниженную чувствительность артериального давления к вливанию ангиотензина II и гиперплазию гранулярных клеток юкстагломерулярного аппарата почки. В результате хронического недостатка калия в организме развиваются слабость или периодический паралич и полиурия. Была описана гиперплазия интерстициальных клеток мозгового вещества почек, вырабатывающих простагландины ПГЕ и ПГF, наряду с повышенной выработкой ПГЕ2. Наследование происходит по аутосомно-рецессивному типу, синдром проявляется в детском возрасте.
Патогенез. Существуют данные о нарушении канальцевого транспорта хлора или калия, хотя патогенетическая последовательность событий до конца не ясна. Любое из этих нарушений может вызвать развитие гипокалиемии, стимулирующей высвобождение простагландинов Е2 и I2 что в свою очередь приведет к повышенной секреции ренина, ведущей к повышенной концентрации циркулирующего в крови ангиотензина II и тем самым альдостерона. Как ангиотензин II, так и альдостерон увеличивают количество почечного калликреина, который вызывает увеличение содержания брадикинина в плазме крови, в то время как альдостерон еще более увеличивает потерю калия через почки. Нормальная ве личина артериального давления поддерживается за счет сосудорасширяющего действия ПГЕ2 и брадикинина, несмотря на увеличенную продукцию ренина, ангиотензина и альдостерона.
Избыточное образование ПГЕ2 в результате гипокалиемии, являющейся стимулятором синтеза ПГЕ2, может быть вторичным следствием этого синдрома. В некоторых случаях блокирование выработки ПГЕ2 индометацином уменьшает уровни содержания ренина в крови и восстанавливает реакцию сосудов на вливание ангиотензина II, но не уменьшает выведение калия из организма.
Лечение. Поступление в организм с пищей количество калия и хлорида натрия ограничивать не следует; могут потребоваться даже добавки калия в пищевой рацион. Посредством фармакологической блокады действия альдостерона на дистальные канальцы (при помощи спиронолактона) можно предотвратить потерю калия из организма, количество же поступающего с пищей натрия следует увеличивать. Как показано выше, применявшееся угнетение синтеза простагландина индометацином, ибупрофеном или ацетилсалициловой кислотой (аспирином) приносило переменный успех. Продуцирование ренина можно снизить блокадой b-адренергических рецепторов.
Синдром Лиддла (псевдогиперальдостеронизм)
Это редко встречающееся наследственное заболевание характеризуется гипертензией, гипокалиемическим алкалозом и малой секрецией альдостерона. Эти нарушения, по-видимому, обусловлены присущей дистальным отделам канальцев или собирательным протокам необычной тенденцией сохранять натрий и экскретировать калий, невзирая на фактическое отсутствие альдостерона. Ни о каких иных биохимических нарушениях не сообщалось. Однако скорости транспорта натрия в эритроциты изменены. Таким больным назначают 100 мг триамтерена в сутки (гл. 182) — мочегонного средства, блокирующего обменные реакции натрия и калия в дистальном канальце.
Дата добавления: 2016-03-05; просмотров: 954;