Конструирование лекарств на основе структур лигандов
(технология LBDD)
Методы LBDD применяются в тех случаях, когда неизвестна пространственная структура мишени. Эти методы основаны на анализе наборов известных лигандов с требуемой биологической активностью. Поскольку структура места связывания лиганда в мишени неизвестна, первоначально необходимо построить его модель. Существует два типа моделей: фармакофорные модели и различные модели «псевдорецептора». Фармакофорная модель представляет собой набор точек в пространстве с определенными физико-химическими свойствами, места связывания и расстояниями между ними. Модели «псевдорецептора» описывают в основном геометрические формы и размер места связывания в мишени. Указанные модели позволяют провести поиск новых лигандов в молекулярных базах данных. Виртуальный скрининг с использованием фармакофорной модели предполагает отбор молекул, удовлетворяющих требованиям данной модели относительно функциональных групп и расстояний между ними. Модели «псевдорецептора» позволяют искать новые лиганды методом молекулярного докинга. Метод «псевдорецептора» предпочтителен в том случае, когда фармакофорная модель содержит относительно мало фармакофорных точек, что приводит к отбору слишком большого количества соединений для экспериментального тестирования.
Существует три основных метода виртуального скрининга потенциальных лигандов:
1) поиск лигандов на основе сходства двухмерных структурных формул;
2) поиск в молекулярных базах данных по фармакофорным моделям;
3) поиск в базах данных методом молекулярного докинга.
Обычно до начала проведения виртуального скрининга выполняется ряд подготовительных процедур - препроцессинг базы данных (удаление из базы соединений, не отвечающих некоторым параметрам: слишком большая или малая молекулярная масса, отсутствие определенных химических групп, несоответствие диапазону физико-химических свойств, таких как гидрофобность, растворимость, проницаемость и др.). Кроме того, часто необходимо конвертировать двухмерные химические структуры в трехмерные, рассчитать конформеры и т.п.
Для предсказания активности отобранных соединений, а также для оптимизации структур базовых соединений используются методы QSAR (Quantitative Structure-Activity Relationship). Это направление возникло на стыке органической химии, математического моделирования и компьютерной химии. Дословный перевод: количественное соотношение структура-свойство. В русском языке для него нет аббревиатуры, поэтому используют английское сокращение.
Исторически всё началось с желания учёных найти количественную связь между структурой вещества и его биоактивностью и выразить её в виде математического уравнения. Это уравнение должно отражать зависимость одного набора цифр (свойств) от другого набора цифр (структур). Однако при этом возникает трудность. Выразить цифрой свойство достаточно просто - физиологическую активность серии веществ можно измерять количественно. Но как выразить числом структуру химического соединения? Над этим вопросом химики и математики работали в течение многих лет. В настоящее время в QSAR используются так называемые дескрипторы химической структуры.
Дескриптор - это число (или математический параметр), которое характеризует структуру органического соединения, причём так, что подмечаются какие-то важные черты этой структуры. В принципе любое число, которое можно рассчитать из структурной формулы - молекулярный вес, число определённых атомов, связей или групп, молекулярный объём, частичные заряды на атомах, - может выступать в качестве дескриптора. Например, годится ли в качестве дескриптора (то есть, характеризует ли соединение) число атомов углерода в нём? Да, и иногда это хороший дескриптор. А число нитрогрупп? Конечно: чем их больше, тем лучше взорвётся - это дескриптор для взрывчатых веществ.
Для предсказания физиологической активности в QSAR обычно используют следующие дескрипторы: электронные эффекты (влияют на ионизацию или полярность соединения), стерические и геометрические особенности структуры (играют важную роль при оценке прочности связывания исследуемого соединения с биомишенью), липофильность (способность растворяться в жирах характеризует способность лекарства преодолевать клеточные мембраны). Большую роль в QSAR имеют так называемые топологические дескрипторы.
В этом методе структурная формула - чисто математическое понятие, граф. Из теории графов можно посчитать так называемые инварианты графов, которые и рассматриваются как дескрипторы. Применяются и сложные фрагментные дескрипторы, которые оценивают вклад различных частей молекулы в общее свойство. Они значительно облегчают исследователям обратное структурное конструирование неизвестных соединений с потенциально высокой активностью. Модель QSAR - это математическое уравнение, с помощью которого можно описать физиологическую активность (и вообще любое свойство).
Метод QSAR работает следующим образом. Сначала группу соединений с известной структурой и известными значениями физиологической активности (полученными из эксперимента) делят на две части: тренировочный и тестовый набор. В этих наборах цифры, характеризующие активность, уже соотнесены с конкретной структурой. Далее выбираются дескрипторы - хорошие компьютерные программы способны перебирать многие сотни дескрипторов. На следующем этапе строят математическую зависимость активности от выбранных дескрипторов для соединений из тренировочного набора и получают так называемое QSAR-уравнение.
Правильность полученного QSAR-уравнения проверяют на тестовом наборе структур. Сначала вычисляют дескрипторы для каждой из тест-структур, подставляют их в QSAR-уравнение, рассчитывают значения активности и сравнивают их с уже известными экспериментальными значениями. Если для тестового набора наблюдается хорошее совпадение расчётных и экспериментальных значений, то данное QSAR-уравнение можно применить для предсказания свойств новых, ещё не синтезированных структур. С помощью этого метода, имея в арсенале совсем небольшое количество химических соединений с известной активностью, можно предсказать необходимую структуру и тем самым резко ограничить круг поисков.
В настоящее время активно применяются методы трехмерного QSAR (3D-QSAR), основанные на описании пространственного распределения свойств лигандов, например, методы сравнительного анализа молекулярных полей (Comparative Molecular Field Analysis, CoMFA) и сравнительного анализа подобия молекул (Comparative Molecular Similarity Indices Analysis, CoMSIA). Эти методы позволяют охарактеризовать области пространства вокруг молекул лигандов (стерические, электростатические, гидрофобные и другие свойства), определяющие взаимодействие макромолекулы-мишени с лимандами из данного набора.
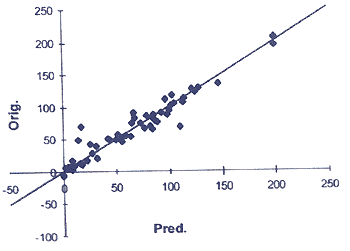 Рис. 6. Пример предсказания активности структур методом QSAR (сравнение экспериментальных и предсказанных данных по ингибированию замещенными индолами захвата Са»+)
Рис. 6. Пример предсказания активности структур методом QSAR (сравнение экспериментальных и предсказанных данных по ингибированию замещенными индолами захвата Са»+)
|
Методы QSAR широко используют химики во всём мире. Например, если взять выпуски журналов „Chemical reviews“ за последние годы, то только в заголовках статей эта аббревиатура встретится несколько раз. Сейчас издаётся несколько специальных журналов, посвященных QSAR.
Дата добавления: 2016-01-30; просмотров: 1261;