Экспериментальное тестирование предсказанных соединений
Технология SBDD дает набор молекулярных структур, ранжированных по неким виртуальным параметрам, но не может ответить на вопрос о возможном биологическом эффекте данных соединений. Поэтому любое компьютерное предсказание необходимо проверять в реальных экспериментах по оценке биологической активности найденных или сконструированных и синтезированных соединений. На первом этапе обычно применяют различные тесты in vitro, которые позволяют быстро оценить способность потенциальных лигандов взаимодействовать с макромолекулой-мишенью. Для этих целей предпочтительно использовать высоко-эффективные системы, основанные на прямом измерении взаимодействия лигандов с мишенями в реальном времени с определением кинетических, равновесных и термодинамических параметров. Кроме того, могут быть использованы разнообразные модели in vivo - от культур клеток до беспозвоночных или «гуманизированных» мышей.
2.4.2. Разработка лекарств на базе соединений – лидеров.
Завершающая стадия создания лекарственного соединения - его разработка. На этой стадии оптимизированный лидер улучшают таким образом, чтобы он стал удобным для клинического использования и приобрёл нужные фармакокинетические характеристики (растворимость в воде, высокую биохимическую стабильность, пролонгированность действия). Как правило, на этой стадии структуру соединений снова изменяют, иногда очень существенно. При этом стараются без крайней необходимости сильно не увеличивать молекулярную массу лекарства, по сравнению с массой молекулы соединения – лидера. Опыт модификации таких сложных природных веществ, как морфин, хинин или алкалоиды кураре свидетельствует, что нередко можно получить более простые аналоги исходного соединения-лидера, сохраняющие его биологическую активность. На этом этапе используется много методов с красивыми названиями: создание биоизостеров, пролекарств, пептидомиметиков, “мягких”(soft drug) и “двойных”(twin drug) лекарств и т. д.
Пролекарства - это вещества, не обладающие выраженной физиологической активностью, но способные превратиться в лекарства уже в организме человека. Происходит это в результате либо ферментативной реакции, либо химической (без участия белкового катализатора). Чтобы получить пролекарство, обычно модифицируют какую-то реакционно-способную группу в физиологически активном соединении так, чтобы эта связь разрушалась в организме. С помощью пролекарств можно, например, продлить действие препарата, повысить его растворимость в воде и даже изменить его вкус.Например,азатиопринявляется пролекарством 6-меркаптопурина, обладающего цитостатическими и иммунодепрессивными свойствами. В организме азатиоприн медленно превращается в 6-меркаптопурин, что приводит к пролонгированию действия последнего.
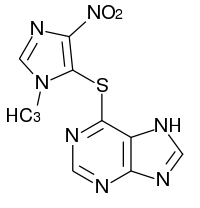
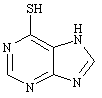
азатиоприн 6-меркаптопурин
Важный методом этого этапа является так называемая изостерическая или биоизостерическая замена. Термин „изостеры“ был введён ещё Ирвингом Ленгмюром в начале XX века: „Молекулы или ионы, которые содержат одинаковое число атомов и имеют одинаковое количество и расположение электронов“. Соответственно изостерическая замена в конструируемом лекарстве - это замена атома или группы на похожую по размеру или валентности. Если при этом сохраняется физиологическая активность, то замена называется „биоизостерической“. Интересно, что термин „биоизостер“ относится и к соединениям, получающимся путём замены на совершенно „непохожие“ группировки, но с сохранением биологических свойств (рис. 7). С помощью биоизостерической замены исследователям удаётся, например, уменьшить токсичность активного соединения, повысить его растворимость и устойчивость к действию ферментативных систем организма и т. д.
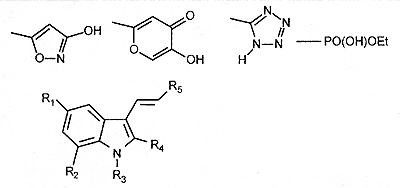 Рис.7. Примеры группировок, которые кажутся „непохожими“ на карбоксильную группу (–СООН), но, тем не менее, часто используются вместо неё при биоизостерической замене
Рис.7. Примеры группировок, которые кажутся „непохожими“ на карбоксильную группу (–СООН), но, тем не менее, часто используются вместо неё при биоизостерической замене
|
Одним из перспективных направлений в создании новых типов лекарственных препаратов является использование соединений, называемых „пептидомиметиками“ и “пептоидами”.
Многие из ферментных субстратов, например ангиотензиноген, ангиотензин, фибриноген (как предшественник фибрина), белки вируса иммунодефицита GAG и GAG-POL (предшественники протеазы и других белков ВИЧ) и многие лиганды ферментов и рецепторов, такие как серпины, энкефалины, нейрокинины, соматостатин, фибриноген, витронектин и другие, являются либо низкомолекулярными пептидами, либо белками. В отличие от взаимодействий белок-белок в сигнальных цепях, взаимодействие этих лигандов с их биомишенями осуществляется с помощью небольшого участка полипептидной цепи, содержащего всего несколько аминокислот, а остальная часть полипептида или белка стабилизирует определенное пространственное расположение данной части макромолекулы. Показательным примером этого может служить пептидный мотив RGD (аргинин, глицин, аспартат), который взаимодействует с различными интегриновыми рецепторами в совершенно разных конформациях.
Если создаваемое нами лекарство должно подействовать на мишень, природный лиганд для которой такой пептид, то этот пептид можно взять в качестве соединения-лидера. Однако пептиды в качестве лекарств не слишком удобны: плохо растворяются в воде, легко расщепляются протеолитическими ферментами организма.
Поэтому на основе такого пептида создается лекарство- пептидомиметик - соединение, способное взаимодействовать с той же мишенью, но содержащее различные непептидные (неприродные) структурные элементы. Пептидомиметики способны копировать или противодействовать биологической активности природных белковых молекул, аналогами которых они являются..
По своей структуре пептидомиметики могут представлять собой полипептидную цепочку, построенную из молекул неприродных α-аминокислот или D-изомеров природных. Введение таких аминокислот (особенно α,α-дизамещенных) в ключевые позиции биологически активных пептидов, как правило, приводит к существенному повышению протеолитической и конформационной стабильности, селективности действия и улучшению фармакокинетических параметров потенциальных лекарств. Пептоидыпредставляют собой полипептиды полученные из молекул глицина, у которых один атом водорода в аминогруппе замещен на различные радикалы.
В принципе, пептидомиметики вовсе не обязательно должны иметь классическую полипептидную структуру. Главное, чтобы они могли эффективно имитировать полипептидный лиганд и обладать высоким сродством к биомишени. Поэтому в структуры пептидомиметиков часто вводят различные фрагменты - заместители и функциональные группы никогда не встречающиеся в природных полипептидах, которые являются биоизостерическими аналогами определенных участков природных пептидов.
Результатом успешного применения пептидомиметического подхода являются некоторые искусственные лиганды интегринов. Сначала была выявлена селективность циклических пептидов по отношению к соответствующим интегриновым рецепторам, а затем уже на их основе были сконструированы бензодиазепиновые пептидомиметики с превосходной селективностью. Другим примером удачного превращения пептидов в пепти-домиметики являются аналоги лигандов рецепторов нейрокинина-1 и нейрокинина-2 и лиганды рецепторов соматостатина с резко выраженной селективностью к определенным их подтипам.
Методом пептидомиметического моделирования также удалось получить ряд ингибиторов протеазы ВИЧ, исходя из последовательности сайта расщепления. Первые лекарства этого типа против ВИЧ - саквинавир, ритонавир и индинавир - еще очень похожи на пептиды, а более новые аналоги, полученные путем структурного дизайна, - нелфинавир, ампренавир, а также пока не выпущенные на рынок ингибиторы, разработанные фирмой «DuPont», уже являются настоящими пептидомиметиками.
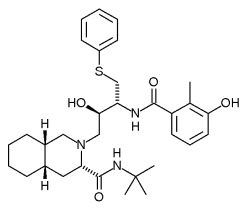
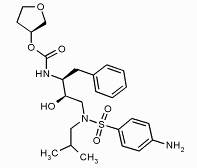
нелфинавир ампренавир
«Мягкие лекарства» - физиологически активные соединения, фармакологический эффект которых локализован в определенном месте. Распределение в других местах приводит к их быстрой деструкции или инактивации. «Мягкие» лекарства представляют собой активные производные неактивных аналогов лекарств. Например, такими являются эфиры кортикостероид-21-карбоновых кислот, которые активны при местном применении, но после всасывания через кожу очень быстро метаболически разлагаются до неактивных 21-кислот. Другим примером являются лекарства против глаукомы.
«Двойные лекарства» - физиологически активные соединения, содержащие две фармакофорные группы, объединенные ковалентно в одну молекулу. Это препятствует образованию молекул соли при комбинации двух лекарств. Примером идентичного двойного лекарства, представляющего собой комбинацию двух одинаковых составляющих может быть симметричная молекула BDHP. Активность BDHP приблизительно в десять раз выше, чем активность составляющих ее молекул нитрендипина (антагониста кальциевых каналов).
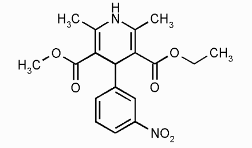
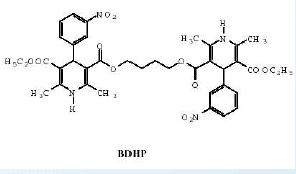
нитрендипин
Двойные лекарства могут быть и неидентичными (имеющими в качестве составляющих различные структуры). В частности, возможно конструирование сложных «бинарных» структур, содержащих в своем составе несколько функционально значимых частей молекулы.
Создание лекарств-клонов.Копирование существующих лекарств с незначительными химическими изменениями (создание лекарств-клонов) получило название исследования «mе too». Такие аналоги иногда могут иметь существенные терапевтические преимущества, по сравнению с уже существующим лекарством. Примером являются, различные биодоступные и устойчивые к лактамазе пенициллины широкого спектра действия, диуретические и противодиабетические сульфамидные препараты на основе антибактериальных сульфамидов. Если лекарство «me too» имеет некоторое терапевтическое преимущество, то оно, как правило, выдвигается на первое место, например, более популярным стал ранитидин по сравнению с циметидином или эналаприл по сравнению с каптоприлом.
Биологическая активность энантиомеров: хиральный переход.В прошлом хиральные лекарства получались в виде рацематов или (при наличии в молекуле нескольких хиральных центров) в виде диастереомерных смесей. Лишь около 20 лет назад фармаколог Арене подверг рацематы критике как вещества, «содержащие 50% примесей». Это подтолкнуло фармацевтическую промышленность к осознанию проблемы, связанной с тем, что оптически активное лекарство и его зеркальный изомер могут значительно различаться по биологической активности. Действительно оказалось, что некоторые хиральные барбитураты в одной из форм обладают седативной активностью, тогда как другой их энантиомер вызывает судороги. В случае синтетических аналогов морфина один энантиомер может быть сильным анальгетиком, а другой - противокашлевым средством. Некоторые дигидропиридины в одной энантиомерной форме являются блокаторами кальциевых каналов, в то время как другой оптический изомер стабилизирует кальциевый канал в открытом состоянии, так что в результате биологические эффекты в таком рацемате компенсируются. Другой пример дает печально известный препарат талидомид. Два его энантиомера обусловливают соответственно седативную активность и побочное тератогенное действие, однако разделение рацемата не приводит к продуктам с индивидуальной активностью из-за метаболической взаимотрансформации обоих энантиомеров.
В течение последнего десятилетия компаниям удалось продлить «время жизни» своих хиральных лекарств с помощью так называемого «хирального перехода», когда на рынок поступает биологически активный энантиомер вместо рацемата. Примерами подобной стратегии являются дексфен-флурамин (изъят из продажи в 1997 г.), дексибупрофен, декскетопрофен, левофлоксацин, левалбутерол, левобупивакаин, эзомепразол, левоцетиризин, дексметилпенидат и эсциталопрам.
«Спасение» плохих соединений-лидеров: метаболический переход.Иногда соединения-лидеры имеют настолько плохие свойства, что ни классическая оптимизация, ни получение пролекарства не в состоянии повысить их терапевтические возможности. Тем не менее, такие соединения можно «спасти», если понятны их биохимические механизмы действия, путем выбора их метаболического предшественника либо активного метаболита неактивного или токсичного лекарства. Приведены ниже отдельные успешные примеры, подтверждают, что «плохие» соединения-лидеры действительно можно трансформировать в ценные лекарства.
Так фенацетин десятилетиями использовался как мягкое анальгетическое и жаропонижающее средство до тех пор, пока при длительном приеме не была обнаружена его гепато- и нефротоксичность, и это лекарство было изъято из продажи. На смену ему пришел активный метаболит фенацетина - парацетамол, не образующий токсичных продуктов.
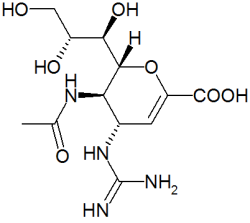
Занамивир - первый ингибитор нейраминидазы, используемый для лечения гриппа, является настолько полярным соединением, что возможно лишь его ингаляционное применение. Анализ химической структуры не привел к изысканию никакого разумного способа его трансформации в орально активную форму лекарства. В то же время случайно обнаруженное сохранение биологической активности у аналогов, не содержащих характерной глицериновой боковой цепи сиаловой кислоты, привело к созданию орально доступного препарата осельтамивира, который представляет собой пролекарство (этиловый эфир) липофильного аналога переходного состояния.
Дата добавления: 2016-01-30; просмотров: 1381;