ГЛАВА 4
|
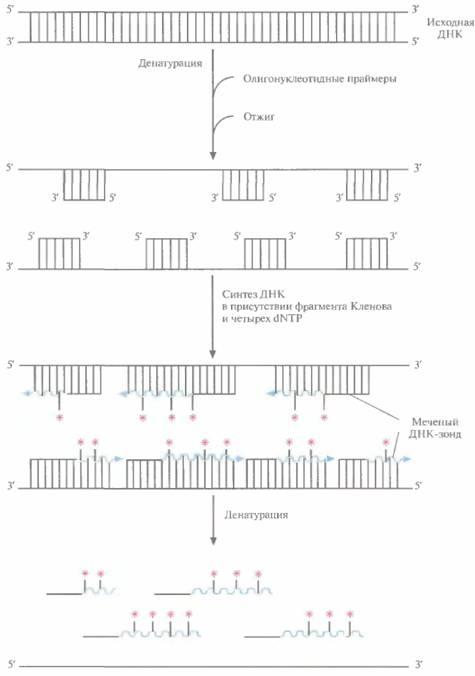
| Рис. 4.12. Получение меченого ДНК-зонда методом случайных праймеров. К денатурированной двухцепочечной ДНК, содержащей нуклеотидную последовательность, которую предполагается использовать в качестве зонда, добавляют гексануклеотиды (смесь всех возможных комбинаций из шести нуклеотидов) и отжигают смесь. Некоторые из олигонуклеотидов гибридизуются с немеченой денатурированной ДНК, и в присутствии фрагмента Кленова и четырех dNTP (один из которых меченый [*]) служат затравкой для синтеза комплементарной цепи. После денатурации синтезированной ДНК получают смесь меченых фрагментов ДНК, которые вместе составляют практически полноразмерную исходную ДНК-матрицу. |
Технология рекомбинантных ДНК 67
ни и гибридизуются с ними, если ДНК предварительно денатурировать (рис. 4.12). После отжига олигонуклеотидов с денатурированной ДНК-матрицей в реакционную смесь добавляют четыре дезоксирибонуклеотида (дезоксирибонуклеозидтрифосфаты; dNTP), один из них — меченый, и фрагмент ДНК-полимеразы 1 Е. соli (фрагмент Кленова). Фрагмент Кленова обладает ДНК-по-лимеразной и 3'-экзонуклеазной активностями, но не 5'-экзонуклеазной активностью, присущей ДНК-полимеразе I E. coli, которая могла бы расщепить новосинтезированные молекулы ДНК, Одиночные цепи ДНК-мишени служат матрицами для синтеза новых молекул ДНК, а связанные с ними случайным образом олигонуклеотиды — затравками (рис, 4,12). При радиоактивном мечении один из dNTP содержит α-32Р, так что 32Р-меченым оказывается и сам зонд. Радиоактивную метку выявляют с помощью радиоавтографии,
В качестве нерадиоизотопной метки часто используют биотин, который присоединяют к одному из четырех dNTP. Для выявления гибридизовавшегося биотинилированного зонда на фильтр наносят конъюгат стрептавидина с соответствующим ферментом (например, щелочной фосфатазой). Стрептавидин образует комплекс с биотином, который обнаруживается благодаря тому, что под действием фермента образуется окрашенное или люминесиирующее вещество — продукт превращения нанесенного на фильтр субстрата.
Зонды для скрининга геномной библиотеки можно получить по крайней мере двумя способами. Во-первых, можно использовать клонированную ДНК близкородственного организма (гетерологичный зонд). В этом случае условия гибридизации нужно подбирать таким образом, чтобы она могла происходить при существенном расхождении между нуклеотидными последовательностями зонда и искомой ДНК; это позволяет решить проблемы, связанные с заведомым различием между ДНК — источником зонда и исследуемой ДНК, Во-вторых, зонд можно получить методом химического синтеза, основываясь на известной аминокислотной последовательности белкового продукта искомого гена.
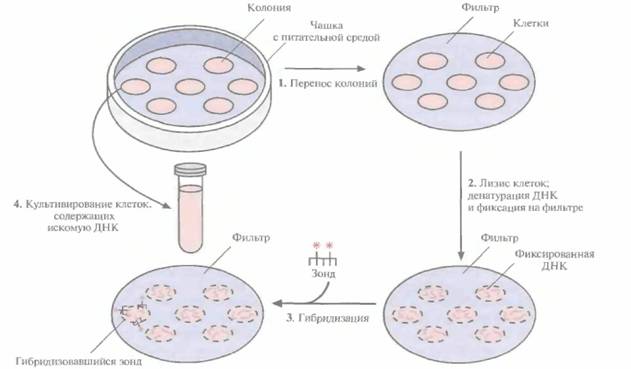
Скрининг библиотек геномных ДНК обычно проводят по следующей схеме. После трансформации высевают клетки на чашки с питательной средой и переносят выросшие колонии на твердую подложку (например, на нитроцеллюлозный или найлоновый фильтр); проводят лизис клеток, затем депротеинизацию и денатурацию ДНК; фиксируют ДНК на подложке. Наносят на фильтр меченый зонд и проводят отжиг, а затем радиоавтографию. Колонии на исходной чашке, которые содержат гибрилизовавшуюся ДНК, выделяют и культивируют (рис. 4.13), Поскольку большинство библиотек получается в результате частичного гидролиза, положительный гибридизационный сигнал может быть получен для нескольких колоний (клонов). Теперь необходимо определить, какой именно клон (если таковой имеется) содержит искомый ген целиком. С помошью гель-электрофореза и картирования определяют размер каждого фрагмента (вставки) и идентифицируют аналогичные фрагменты или фрагменты с перекрывающимися последовательностями. Можно также провести дополнительное клонирование с тем, чтобы составить полный ген из перекрывающихся фрагментов. Или, если вставка в каком-либо из клонов достаточно велика и вполне может содержать весь ген, провести ее секвенирование и убедиться в наличии старт- и стоп-кодонов и полноразмерной нуклеотидной последовательности, кодирующей искомый белок.
К сожалению, никто не может дать гарантии, что в библиотеке представлена вся нуклеотидная последовательность нужного гена. Если поиск полноразмерного гена оказался безрезультатным, можно создать другую библиотеку, используя другую рестриктазу, и провести скрининг с помощью исходного зонда или зондов, созданных на основе предыдущей библиотеки. Чтобы повысить вероятность присутствия в библиотеке полной версии искомого гена, можно также создать библиотеки, содержащие фрагменты ДНК заведомо большего размера, чем средний размер прокариотического гена (этот вариант мы рассмотрим вданной главе позже).
Иммунологический скрининг
В отсутствие ДНК-зонда для скрининга геномной библиотеки можно использовать другие методы. Например, если клонированный ген экс-прессируется, то его продукт — весь белок или его часть — можно обнаружить иммунологиче-
68 ГЛАВА 4
|
| Рис. 4.13. Скрининг библиотеки геномной ДН К с применением меченого зонда. Клетки после трансформации высевают на твердую питательную среду, обеспечивающую рост только трансформированных клеток. 1 . Переносят клетки из каждой выросшей колонии на твердую подложку (например, нитроцеллюлозный или найлоновый фильр) так, чтобы их расположение соответствовало таковому на чашке. 2. Клетки лизируют, высвободившуюся ДНК подвергают денатурации и депротеинизации и фиксируют на фильтре. 3, На фильтр наносят меченый ДНК-зонд и проводят гибридизацию. Смывают с фильтра негибридизовавшийся зонд и проводят радиоавтографию с тем чтобы определить, какие клетки содержат меченый ДНК-зонд. 4, Идентифицируют на чашке колонии, содержащие искомую ДН К (положительный гибридизационный сигнал), отбирают из них материал и культивируют. |
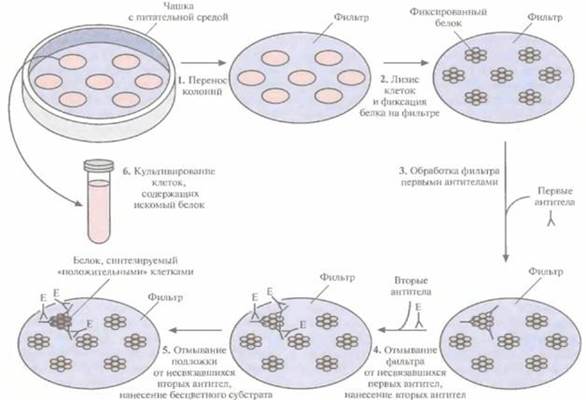
скими методами. Технически эта процедура имеет много общего с гибридизацией. Все клеточные линии (клоны) библиотеки высевают на чашки с питательной средой. Выросшие колонии переносят на фильтр, клетки лизируют, а высвободившиеся белки фиксируют на фильтре. Затем на фильтр наносят первые антитела, которые специфически связываются с данным белком (антигеном), все несвязавшиеся антитела удаляют, а фильтр помещают в раствор вторых антител, специфичных в отношении первых антител. Во многих тест-системах используют конъюгаты вторых антител с ферментом, например с щелочной фосфатазой. После отмывания фильтра добавляют бесцветный субстрат. Если вторые антитела связываются с первыми, то под действием фермента происходит гидролиз субстрата с образованием окрашенного вещества в том месте, где идет реакция (рис. 4.14).
Те клетки на чашке, которые соответствуют окрашенным пятнам на фильтре, содержат или полноразмерный ген, или достаточно протяженный его участок, обеспечивающий синтез белкового продукта, узнаваемого первыми антителами. По окончании иммунологического скрининга геномной библиотеки необходимо определить, какой именно из отобранных клонов содержит полноразмерный ген.
Технология рекомбинантных ДНК 64
|
| Рис. 4.14. Иммунологический скрининг геномной библиотеки (иммунологическое тестирование колоний). Клетки после трансформации высевают на твердую питательную среду, обеспечивающую рост только трансформированных клеток. L Переносят клетки из каждой выросшей колонии на твердую подложку (например, нитроцеллюлозный или найлоновый фильтр) так, чтобы их расположение в точности соответствовало таковому на чашке. 2. Клетки подвергают лизису, белки фиксируют на фильтре. 3. Наносят на фильтр первые антитела, которые связываются только с искомым белком. 4. Несвязавшиеся первые антитела удаляют, наносят на фильтр вторые антитела, специфичные в отношении первых антител, связанные с ферментом (например, щелочной фосфатазой). 5. Смывают с фильтра все несвязавшиеся вторые антитела и наносят на него бесцветный субстрат, при гидролизе которого образуется окрашенный продукт. Гидролиз может произойти только в присутствии вторых антител. 6. Отбирают колонии на чашке, соответствующие окрашенным пятнам на фильтре, и культивируют их. В них может содержаться рекомбинантная ДНК, кодирующая белок, гомологичный первым антителам. |
Скрининг по активности белка
Метод гибридизации ДНК и иммунологические методы позволяют идентифицировать многие гены и их продукты. Если при этом искомый ген кодирует фермент, не синтезируемый клеткой-хозяином, то для обнаружения клонов, содержащих данный ген, можно использовать метод идентификации на чашках. Так были идентифицированы гены α-амилазы, эндоглюканазы и ß-галактозидазы различных организмов. Для этого клоны Е. соli, составляющие геномную библиотеку данных организмов, высевали на чашках с питательной средой, содержащей специфический субстрат. После окрашивания селективным красителем клетки, способные утилизировать этот субстрат, приобретали характерную окраску.
70 ГЛАВА 4
Если искомый ген кодирует продукт, без которого мутантная клетка-хозяин не может расти на минимальной среде, то библиотеку можно создать методом трансформации мутантных клеток. Клетки, выросшие в отсутствие необходимого субстрата на минимальной среде, обязательно содержат функциональный искомый ген, попавший в клетку в составе плазмидного вектора. В разных вариантах этот подход использовали для выделения многих важных генов, в частности генов, ответственных за синтез антибиотиков и образование азотфиксирующих клубеньков на корнях некоторых растений,
Клонирование структурных генов эукариот
Для клонирования эукариотических структурных генов необходимы специальные методики. Прокариоты не способны удалять интроны из первичных РНК-транскриптов, поэтому правильная трансляция эукариотических мРНК в бактериальной клетке невозможна. Более того, экспрессия эукариотической ДНК может осуществляться только при наличии прокариотических сигнальных последовательностей, регулирующих транскрипцию и трансляцию. Концевые участки эукариотических мРНК особым образом модифицированы: их 5'-концы кэпированы (содержат «кэш из остатка G, часто метилированного), а 3'-концы полиаденилированы (содержат poly(A)-«хвост» из примерно 200 остатков аденозина).
Наличие роlу(А)-хвоста позволяет отделить мРНК от рибосомной и транспортной РНК. Для этого суммарную эукариотическую РНК пропускают через колонку, заполненную целлюлозой, к которой «пришиты" короткие олигонуклеотидные цепочки из тимидиновых остатков длиной примерно 15 звеньев, oligo(dT). Ро)у(А)-хвосты молекул мРНК спариваются с oligo(dT) и задерживаются в колонке, а молекулы тРНК и рРНК свободно проходят через нее. Затем колонку промывают буфером, в котором происходит разрыв водородных связей между А и Т, и мΡΗΚ высвобождается.
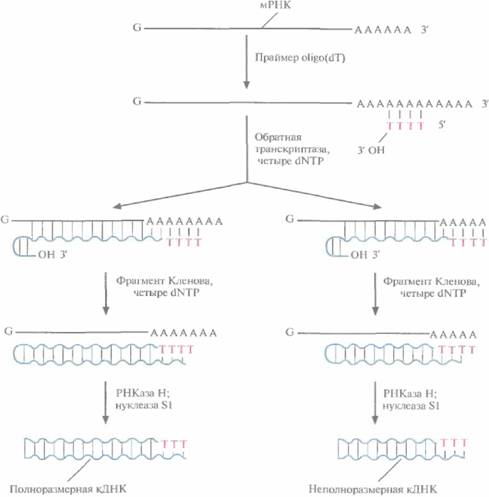
Саму мРНК нельзя встроить в ДНК-вектор, сначала на ней необходимо синтезировать двух-цепочечную ДНК, Для этого последовательно используют две разные полимеразы: обратную транскриптазу и фрагмент Кленова ДНК-поли-меразы I (рис. 4.15). Вначале в реакционную смесь с очищенной мРНК добавляют короткие oligo(dT), обратную транскриптазу и четыре dNTP (dATP, dTTP, dGTP, dCTP). Ро1у(А)-хвост мРНК спаривается с oligo(dT), несушим свободную 3'-ОН-группу, которая инициирует синтез комплементарной цепи. Матрицей в этом синтезе служит молекула мРНК, а катализирует его обратная транскриптаза, продуцируемая некоторыми РНК-вирусами. Она последовательно присоединяет к растущей цепи остатки Т, С, G или А, комплементарные A, G, С или U мРНК. In vitro синтез ДНК идет не до конца, при этом обратная транскриптаза перед остановкой обычно «поворачивает вспять» и присоединяет несколько нуклеотидов в обратном направлении (рис. 4.15), так что в результате образуется «шпилька».
В реакционную смесь добавляют фрагмент Кленова ДНК-полимеразы I E. coli, который достраивает вторую цепь ДНК, используя первую цепь как матрицу. Он присоединяет дезоксирибонуклеотиды к растущей цепи, начиная с 3'-ОН-конца шпильки. По окончании синтеза препарат обрабатывают ферментом PHКазой Η, которая разрушает молекулы мРНК, и нуклеазой SI, отщепляющей одноцепочечные концы ДНК. Полученный препарат представляет собой смесь частично и полностью двухцепочечных комплементарных ДНК-копий (кДНК) мРНК, преобладающей в исходном образце.
Разные кДНК можно встроить в плазмидный вектор и получить кДНК-библиотеку. Для скрининга кДНК-библиотеки с целью идентификации клонов, несущих специфические гибридные плазмиды, можно использовать метод гибридизации или иммунологические методы, В последнем случае кДHК должна быть встроена в сайт, находящийся под контролем бактериального промотора, обеспечивающего транскрипцию. Однако практически ни один вектор не гарантирует, что во встроенной кДНК сохранится правильная рамка считывания и синтезируется правильная полипептидная цепь. Тем не менее все положительные клоны, выявленные тем или иным методом, необходимо подвергнуть дальнейшей проверке и идентифицировать те из них, ко-
Технология рекомбинантных ДНК 71
|
| Рис. 4.15. Синтез кДНК. К препарату очищенной мРНК добавляют праймер oligo(dT). Для синтеза ДНК на РНК-матрице используют фермент обратную транскриптазу и четыре dNTP. I n vitro обратная транскриптаза не обеспечивает синтез полноразмерных кД H К- коп и и на всех матрицах и образует на конце растущей цепи шпильку со свободной 3'-ОН -группой. Эта группа инициирует синтез второй цепи ДНК при участии фрагмента Кленова. После завершения синтеза молекулы мРНК гидролизуют РНКазой Н, а ДНК обрабатывают нуклеазой SI , в результате чего получаются линейные молекулы ДНК с тупыми концами без шпилек. |
торые несутполноразмерную нуклеотиднуюпоследовательность, кодирующую белок-мишень.
Векторы для клонирования крупных фрагментов ДНК
Векторы на основе бактериофага λ
С помощью плазмидных векторов можно клонировать фрагменты ДНК длиной до 10 т. п. н. Однако при создании геномных библиотек часто приходится работать с более крупными фрагментами. Для этого были разработаны векторы на основе бактериофага λ Ε. coli.
После проникновения фага λ в клетку E. coli события могут развиваться по двум сценариям. Если реализуется литический цикл, то фаг начинает интенсивно размножаться и примерно через 20 мин клетка разрушается (лизирует) с высвобождением до 100 новых фаговых частиц. При альтернативном варианте развития событий фаговая
72 ГЛАВА 4
ДНК включается в хромосому Е. coli как профаг и реплицируется в клетке вместе с нормальными бактериальными генами (состояние лизогении). Однако при недостатке питательных веществ или иных неблагоприятных обстоятельствах интегрированная фаговая ДНК высвобождается, и запускается литический цикл развитии. Размер ДНК фага λ составляет примерно 50 т. п. н., причем значительная ее часть (около 20 т. п. н.) несущественна для размножения фага и отвечает за его встраивание в хозяйскую ДНК. В связи с этим возникла идея, что ее можно заменить фрагментом другой ДНК эквивалентного размера. Образующаяся ре-комбинантная молекула будет реплицироваться в клетке как ДНК «рекомбинантного" фага λ, «вставшего" на литический путь развития.
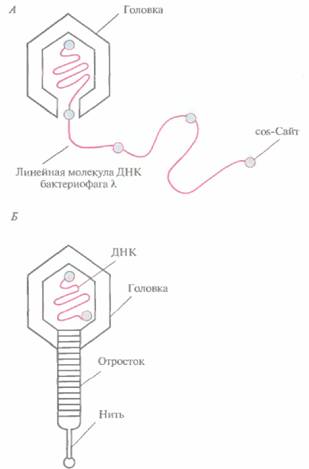
Чтобы понять, как функционирует векторная система на основе фага λ, необходимо рассмотреть молекулярные аспекты литического цикла развития. Инфекционная фаговая частица имеет головку, в которой заключена плотно упакованная ДНК длиной примерно 50 т. п. н., и отросток с отходящими от него тонкими белковыми нитями (фибриллами). Сборка головки и отростка и упаковка ДНК четко скоординированы. ДНК фага λ — это линейная двухцепочечная молекула длиной 50 т. п. н. с одноцепочечными 5'-«хвостами» из 12 нуклеотидов. Их называют липкими (cos) концами, поскольку они взаимно комплементарны и могут спариваться друг с другом. После того как фаговая ДНК проходит через отросток и попадает в E. coli, cos-концы соединяются с образованием кольцевой молекулы. На раннем этапе литического цикла в результате репликации кольцевой молекулы ДНК образуется линейная молекула, состоящая из нескольких сегментов длиной 50 т. п. н. (рис. 4.16, A). Каждый из таких сегментов упаковывается в белковую головку, к последней присоединяется уже собранный отросток и образуется новая фаговая частица (рис. 4.16,Б). При упаковке молекулы ДНК длиной менее 38 т. п. н. получается неинфекционная фаговая частица, а фрагменты длиной более 52 т, п. н. не умещаются в головку. Сегменты длиной 50 т. п. н. в линейной молекуле ДНК разделены cos-сайтами, и именно по этим сайтам разрезается молекула, когда очередной сегмент заполняет головку. Разрезание осуществляет фермент, находящийся у входа в головку.
В результате исследований по изучению сборки фага λ была разработана система упаковки молекул ДНК in vitro с образованием инфекционных фаговых частиц. Смешав в пробирке очищенные пустые головки, фаговую ДНК и собранные отростки, можно получить инфекционные фаговые частицы.
Один из множества λ-векторов для клонирования имеет два BamHI-сайта, фланкирующих участок длиной 20 т. п. н. При гидролизе очищенной фаговой ДНК рестриктазой ВатНIобразует-
|
| Рис. 4.16. Литический путь развития бактериофага λ. А. При репликации кольцевой ДНК. бактериофага λ образуется линейная молекула, состоящая из повторяющихся сегментов длиной примерно 50 т. п. н. Каждый из этих сегментов представляет собой полноразмерную фаговую ДНК. Б, Фаговая головка вмещает один такой сегмент, затем к головке присоединяется уже собранный отросток. |
Технологиярекомбинантных ДНК 73

|
| Рис, 4.17. Клонирующая система на основе бактериофага λ. Фаговая ДНК имеет два BamHI-сайта, фланкирующих ее I/Е-сегмент. Клонируемую ДНК расщепляют с помощью BamHI, фракционируют полученные фрагменты по размеру и выделяют те из них, которые имеют размер от 15 до 20 т.п.н. Фаговую ДНК обрабатывают этим же ферментом. Оба препарата ДНК смешивают и обрабатывают ДНК-лигазой фага Т4. Лигированная смесь содержит самые разные комбинации ДНК, в том числе 1) восстановленную ДНК фага λ и 2) рекомбинантные молекулы, содержащие R- и L-области фаговой ДНК и вставку клонируемой ДНК размером ~20 т. п. н, занявшую место области I/Е фагового генома. Рекомбинантные молекулы упаковывают в головки бактериофага λ in vitro, и после добавления отростков получают инфекционные фаговые частицы. В инфицированных рекомбинантным фагом клетках Е. соli, в хромосому которых интегрирована ДНК бактериофага Р2, могут реплицироваться и образовывать инфекционные частицы только молекулы ДНК, составленные из R- и L-областей фаговой ДНК и клонированной вставки размером ~20 т.п.н. |
Дата добавления: 2015-07-14; просмотров: 1506;