Циклы большего размера. Принципы макроциклизации. Эффекты многоцентровой координации
| Число атомов в цикле (п) | Относительная скорость(при 50˚С) |
| 1,5 10е | |
| 1,7 • 104 | |
| 97,3 | |
| 1,00 | |
| 1,12 | |
| 3,35 | |
| 8,51 | |
| 10,6 | |
| 32,2 | |
| 41,9 | |
| 45,1 | |
| 52,0 | |
| 51,2 | |
| 60,4 |
Для реакций ациклических бифункциональных предшественников, результатом которых может быть образование циклов большего размера, по мере возрастания числа звеньев вес менее вероятным становится образование циклического переходного состояния. Это прежде всего связано с возрастанием энтропийного барьера для образования свернутой конформации ациклической молекулы [30а]. Сказанное можно продемонстрировать приведенными ниже данными по сравнительным скоростям превращения серии бромэфиров 305в соответствующие лактоны 306(схема 2.113) [ЗОЬ]:
По мере снижения скорости лактонизации все в большей степени преобладающим становится межмолекулярная конденсация с образованием оли-гомерных сложных эфиров. Так, например, практически невозможно получить десятичленный лактон 307из предшественника 308в условиях, оптимальных для получения пяти- или шестичленных циклов; основным продуктом при этом неизменно будет олигомер 309.
Изменить ход событий в желательном направлении можно, очевидно, двумя способами: путем селективного подавления межмолекулярной реакции либо путем форсирования внутримолекулярной. Однако сложность задачи состоит в том, что по своему химизму эта две реакции тождественны. Поэтому разобранные нами ранее принципы управления селективностью реакции в данном случае не могут быть эффективными. Тем не менее эта задача оказалась разрешимой.
 Схема 2.113
Схема 2.113
|
Классический метод проведения макролактонизации был разработан в группе Цитлера в 1930-х годах [30с]. Суть этого метода — использование условий высокого разбавления. В этих условиях резко уменьшается вероятность межмолекулярных столкновений и соответственно подавляется образование олигомерных продуктов. В то же время скорость внутримолекулярной реакции не зависит от концентрации субстрата, которая в общем случае не может повлиять на вероятность встречи двух концов одной и той же молекулы. Этот метод вполне универсален, и с его помощью в 1930—50-х годах были выполнены многочисленные синтезы соединений, содержащих циклы среднего и большого размеров [30с]. Тем не менее явные технические неудобства этого метода (малые количества вещества при большом количестве растворителя) требовали разработки альтернативных путей, основанных на избирательном форсировании внутримолекулярной реакции.
Проблема макролактонизации встаца с особой остротой в 1960-х годах, когда начались интенсивные работы по полному синтезу природных антибиотиков, содержащих в своем составе макроциклические лактонные циклы (макролиды). В результате серии углубленных исследований проблему создания препаративно удобных методов получения макроциклических лакто-нов с почти любым размером цикла удалось решить [30d].
Один из наиболее успешных подходов основан на принципе двойной активации по обеим концам циклизуемого субстрата, как это показано на схеме 2.114 для общего случая превращения оксикислот типа 310в лактоны 311.На начальной стадии оксикислота 310превращается в соответствующий 2-пиридинотиоэфир 312.Стадия лактонизации проводится путем прибавления тиоэфира 312в кипящий ксилол. При этом с хорошим выходом и без использования высокого разбавления могут быть получены макролактоны, в том числе и для п = 10 - 14 (наиболее часто встречающийся размер цикла в макролидах).
 Схема 2.114
Схема 2.114
|
По-видимому, наблюдаемая предпочтительность лактонизации по сравнению с межмолекулярной этерификацией обусловлена возможностью промежуточного образования бетаиновых производных 312аи 312Ь(за счет внутримолекулярного переноса протона), что существенно ускоряет внутримолекулярную циклизацию (для описания эффекта предложен термин — «электростатическое стимулирование») [ЗОе]. Иллюстрацией эффективности этого подхода может служить синтез природного макролида ресифейолида (313),содержащего 12-членный цикл, из оксикислоты 314с выходом 52% [30f].
Совершенно иной подход к решению проблемы обеспечения эффективности внутримолекулярной циклизации появился благодаря пионерским исследованиям Педерсена [2с] по синтезу краун-эфиров. Действительно, уже в одной из первых его работе сообщалось об удивительном факте, а именно об образовании с высоким выходом 18-членного полиэфира 315при взаимодействии 2 экв. пирокатехина (316)с 2 экв. бис-(2-хлорэтилового) эфира (317)(схема 2.115). Удивительном было то, что в этом синтезе макроцикла вовсе не требовалось высокого разбавления. Действительно, получение 1 моля (360 г) продукта 315потребовало использования всего лишь 5 л растворителя!
 Схема 2.115
Схема 2.115
|
Наблюдаемая эффективность реакции была объяснена матричным эффектом иона натрия, который благодаря образованию координационных связей с атомами кислорода способен эффективно стабилизировать квазициклическую конформацию субстрата 318 на стадии циклизации. Тем самым обеспечивается принудительное сближение реагирующих центров, резко облегчающее образование циклического продукта — натриевого комплекса 315а. Декомплексация последнего и дает 18-членный краун эфир 315. Та же реакция при попытке ее проведения в присутствии гидроксида лития или аммония не дает 315, а приводит к образованию ациклических олиго-мерных эфиров. Последующие исследования надежно подтвердили справедливость концепции многоцентрового связывания, и на этой основе развилась самостоятельная область органической химии, о которой мы более подробно поговорим в гл. 4.
Концептуально сходный подход, основанный, однако, на другом типе связывания, был успешно применен в синтезе некоторых макроциклических алкалоидов, как это показано на схеме 2.116. Лактам 319, содержащий 13-членный цикл, является основным структурным фрагментом алкалоида целасинина. В исследованиях Ямамото с сотр. [30g] в качестве наиболее естественного предшественника для синтеза 319 был избран триамин 320, поскольку синтез последнего был легко осуществим, а его превращение в 319 требовало «всего лишь» внутримолекулярного образования амидной связи. Однако сложность задачи состояла в том, что требовалось обеспечить региоселективность внутримолекулярного аминолиза сложноэфир-ной группы с участием только терминальной аминогруппы и сделать мак-роциклизцию более предпочтительным направлением реакции, чем олиго-меризация. Очевидно, что для решения этих задач необходимо было каким-то образом стабилизировать требуемую циклическую конформацию субстрата 320. В данном случае этого удалось добиться благодаря использованию бора в качестве временного связующего звена.
Действительно, было хорошо известно, что производные бора легко образуют координационные связи с аминами и способны превращаться в соединения с ковалентной связью В-Н, которая может подвергаться гидролитическому расщеплению в слабокислых условиях. Ближайшей моделью требуемого превращения могла служить ранее описанное превращение триамина 321 в триазаборабициклсщекан (323) под действием три с-(ди метилами но)борана 322. Оказалось, что в практически тех же самых условиях можно провести циклизацию 320 с образованием бициклического интермедиата 320а. Жесткая структура последнего, очевидно, обеспечивала требуемое сближение этокси-карбонильной и аминной групп, поскольку стадия образования лактамной связи спонтанно протекала в этих же условиях, и обработка реакционной массы хлоридом аммония дала продукт 319 с выходом 77% [30g].
Подчеркнем, что в последнем случае, так же как и при синтезе краун-эфира 315, не было нужды прибегать к технике высокого разбаачения, так как требуемый результат — предпочтительность внутримолекулярного пути — обеспечивался благодаря фиксации требуемой геометрии субстрата за счет вспомогательных факторов, играющих роль, так сказать, «организующего начала». Ниже мы еще не раз сможем убедиться в том, насколько эффективным может быть подобный прием при решении синтетических задач самого различного рода.
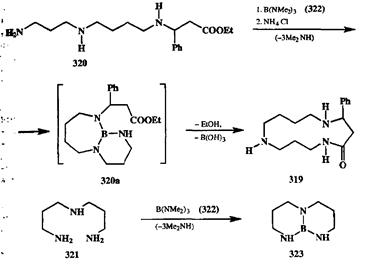 Схема 2.116
Схема 2.116
|
Дата добавления: 2015-04-05; просмотров: 1223;