Термодинамический и кинетический контроль
Для того чтобы термодинамически допустимое превращение X → Y могло осуществиться, реагирующая система X (это может быть одно вещество или несколько компонент, словом, все участники процесса), как правило, должна преодолеть некоторый потенциальный барьер. Возникновение этого барьера обусловлено необходимостью для системы пройти переходное состояние, которое более богато энергией, чем исходные или конечные продукты. Энергией, достаточной для преодоления барьера, обладает за счет флуктуации лишь небольшая часть сталкивающихся молекул, и лишь малая часть столкновений происходит при нужной для реакции взаимной ориентации молекул. Поэтому органические реакции протекают не мгновенно, а с измеримой скоростью, величина которой зависит от высоты барьера (энергии активации). Если барьер мал, то скорость реакции высока, а если очень велик, то скорость реакции почти нулевая. Наличие подходящего канала для реакции или, что тоже, существование подходящего механизма для данного превращения означает наличие возможности реализации переходного состояния сравнительно низкой энергии (рис.2.2).
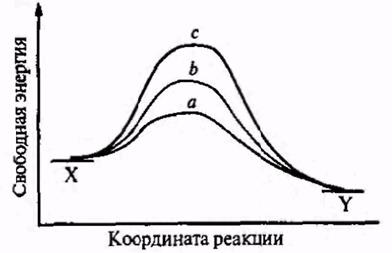 Рис.2.2. Потенциальные барьеры реакции: а — барьер мал, реакция протекает почти мгновенно; B — барьер средней высоты, реакция протекает с измеримой скоростью; с— очень высокий барьер, реакция практически не идет.
Рис.2.2. Потенциальные барьеры реакции: а — барьер мал, реакция протекает почти мгновенно; B — барьер средней высоты, реакция протекает с измеримой скоростью; с— очень высокий барьер, реакция практически не идет.
|
-> Вообще говоря, барьер между исходными веществами и продуктами реакции никогда не бывает бесконечно высоким. В самом деле, у нас всегда есть в запасе теоретически мыслимый крайний путь, определяющий верхний предел высоты барьера, а именно: разобрать исходные молекулы на атомы (например, пиролизом), а потом собрать их по-новому так, чтобы образовались нужные п^родукты. При условии, что это превращение термодинамически разрешено, то и этот путь мог бы рассматриваться как вполне законный, если бы не существовало никаких других разрешенных направлений превращения системы.
Рассмотрим ту же систему X →Y, прибавив еще один компонент — термодинамически более стабильный продукт Z (рис. 2.3).
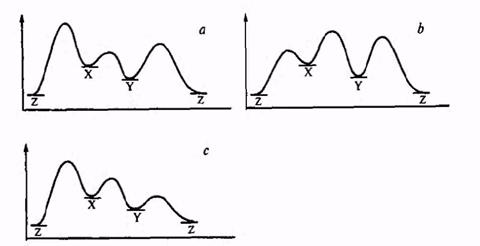 Рис.2.3. Энергетические профили реакций с кинетическим или термодинамическим контролем: а — реакция X → Y доминирует, продукт Y кинетически устойчив; B — доминирует побочная реакция X→Z;c — доминирует реакция X → Y, но продукт Y кинетически неустойчив и быстро превращается в побочный продукт Z.
Рис.2.3. Энергетические профили реакций с кинетическим или термодинамическим контролем: а — реакция X → Y доминирует, продукт Y кинетически устойчив; B — доминирует побочная реакция X→Z;c — доминирует реакция X → Y, но продукт Y кинетически неустойчив и быстро превращается в побочный продукт Z.
|
Наиболее благоприятный случай для синтеза Y представлен кривой а. Исходная система X может реагировать в двух направлениях: с образованием целевого продукта Y и побочного продукта Z. Барьер первой реакции существенно ниже, чем второй. Поэтому скорость реакции образования целевого продукта значительно превышает скорость побочной реакции, в результате чего весь исходный материал X расходуется на образование Y, а побочная реакция просто не успевает пройти. Хотя по условиям задачи Z является более стабильным продуктом, в рассматриваемом случае его образование не происходит ш-за достаточно высокого барьера для превращения Y -* Z. Стабильность Y есть стабильность кинетическая, и возможность его образования из X в данном случае определяется низкой скоростью превращения Y в термодинамически более выгодный продукт Z. Иными словами, Y по-прежнему сохраняет тенденцию «свалиться в яму», но система «заперта» потенциальными барьерами, которые и обеспечивают возможность ее сохранения. Именно наличие сравнительно высоких потенциальных барьеров обусловливает возможность существования огромного числа органических соединений, подавляющее большинство которых термодинамически нестабильны, а также и возможность проведения синтеза «против термодинамики», как в рассмотренном на рис. 2.1 примере получения продукта Р из исходного соединения А.
Кривые b и с описывают неблагоприятные ситуации для синтеза Y из X. В первом из этих случаев преобладает побочная реакция X -> Z, и продукт Y просто не успевает образоваться. Во втором случае желаемая реакция осуществляется достаточно легко, но продукт Y оказывается кинетически нестабильным, и из-за малости барьера между Y и Z превращение Y -> Z протекает очень легко, и в этом случае образование продукта Z и будет основным результатом превращения системы X.
Следующие два конкретных примера могут служить хорошей иллюстрацией роли кинетической и термодинамической стабильности. Как известно, в середине XIX в. острейшие дискуссии вызывала структура бензола — соединения, которому долгое время не удавалось приписать непротиворечивую структурную формулу. В числе других вариантов в 1867 г. Дьюаром была предложена для бензола гипотетическая структура 2. Довольно быстро быто показано, что эта структура неверна и на самом деле наилучшим представлением строения бензола следует считать предложенную Кекуле структуру 3. Однако почти столетие спустя оказалось, что структура 2, предложенная Дьюаром, не так уж фантастична, и соединение, имеющее такое строение, «бензол Дьюара», удалось получить ван Тамелену [1а]. Как и следовало ожидать, соединение 2 оказалось гораздо менее стабильным термодинамически, чем «нормальный» бензол 3 (схема 2.2). Действительно, превращение 2 → 3 протекает с выделением значительной энергии, примерно 14—17 Дж/моль. Тем не менее, 2 оказался довольно устойчивым соединением даже при комнатной температуре, и его самопроизвольная изомеризация в 3 в этих условиях протекает довольно медленно (время полупревращения при 20°С составляет примерно 2 сут). Эти наблюдения указывают на существование достаточно высоких энергетических барьеров, буквально запирающих молекулу 2 в потенциальной яме и предотвращающих ее немедленное превращение в бензол. Естественно, что синтез такого соединения оказался возможным только потому, что для этого был разработан путь, исключающий все возможные альтернативы, которые могли бы привести к образованию более стабильного 3.
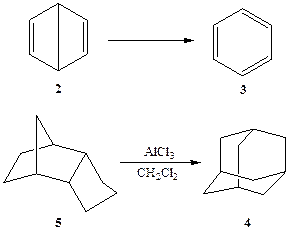 Схема 2.2
Схема 2.2
|
В некотором смысле альтернативным примером может служить адамантан (4). Этот углеводород, скелет которого представляет собой повторяющийся элемент кристаллической структуры алмаза, вплоть до 40-х годов XX столетия еще считался довольно экзотическим веществом, поскольку его многостадийный синтез оставался слишком трудоемким делом. Впоследствии было установлено, что среди всех возможных изомеров состава C]0Hi6 именно структура адамантана отвечает минимуму потенциальной энергии. Подобная исключительная термодинамическая стабильность 4 подсказала возможность его синтеза путем термодинамически контролируемой изомеризации других углеводородов СюН^. Действительно, как было показано Шлейером [1Ь], легко доступный углеводород 5 может претерпевать под действием кислотных катализаторов превращение в 4. Впоследствии эта реакция была усовершенствована и стала удобным препаративным методом получения адамантана (4). Тот факт, что адамантан обнаруживается в заметных количествах в ископаемом углеводородном сырье (в некоторых сортах нефти), также является следствием и наглядной демонстрацией его высокой термодинамической стабильности.
Необходимо подчеркнуть, что энергетические профили типа показанных на рис. 2.2 и 2.3, не есть нечто абсолютное, жестко задаваемое только структурой рассматриваемых соединений. Огромную роль в реализации той или иной энергетической картины играют внешние условия, в которых проводятся превращения: природа растворителя и температура, наличие и природа катализаторов, темновые условия или облучение и т. п. Влияние этих факторов на возможность реализации того или иного пути реакции в общем поддается рациональной интерпретации. Соответственно, при правильном выборе параметров в очень многих случаях удается обеспечить протекание реакции в направлении, требуемом для решения данной синтетической задачи.
Рассмотрим, насколько разнообразными могут быть пути превращений одного органического соединения и какими способами удается управлять направленностью этих превращений на примере реакций толуола (6) (схема 2.3).
Две из показанных на схеме реакций толуола, а именно (1) и (2) проходят по одной и той же стехиометрической схеме:
С7Н8 + Вг2 → С7Н7Вг + НВг
но дают изомерные продукты — бензилбромид (7) и п-бромтолуол (8). В подходящих условиях получение каждого из них можно провести избирательно при практически полном подавлении конкурирующего процесса. Для того чтобы понять, каким образом этого удается добиваться, необходимо проанализировать механизм этих превращений.
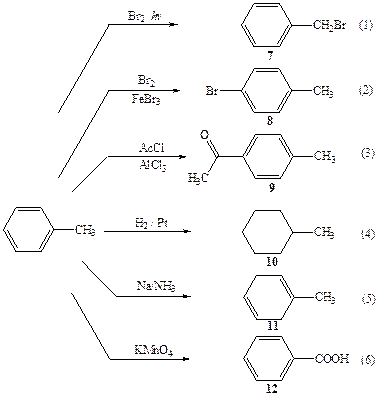 Схема 2.3
Схема 2.3
|
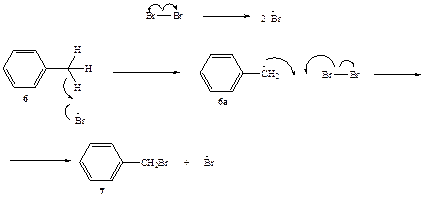 Схема 2.4
Схема 2.4
|
Механизм бромирования толуола, приводящий к образованию бензил-бромида (7), показан на схеме 2.4. Истинным реагентом, атакующим молекулу толуола, является атомарный бром, образующийся при облучении за счет обратимой диссоциации молекулы брома под действием фотона. В атоме брома заключается около 50% энергии фотона, поглощенного молекулой Вг3, поэтому он является высоко активной частицей, способной отрывать атомы или радикалы от других молекул. Хотя в молекуле толуола содержится 8 атомов водорода, только три из них, а именно водороды метальной группы, способны эффективно реагировать с атомарным бромом. Причина избирательности состоит в том, что результатом такой реакции будет образование бензилъного радикала (6а), наиболее стабильного из радикалов, которые мо-i^r образоваться из толуола. Бензильный радикал далее взаимодействует с молекулярным бромом, давая конечный продукт 7 и регенерируя при этом атом брома, что и обеспечивает возможность продолжения цепи реакций.
Реакция толуола с бромом в присутствии бромида железа(Ш) протекает по совершенно другому механизму. Здесь реагентом не является ни молекулярный, ни атомарный бром. В этом случае молекулярный бром при действии бромида железа(Ш) превращается в солеобразный комплекс, условно обозначаемый как Br+[FeBr4]- (схема 2.5). Эта реакция является обратимой, и стационарная концентрация бром-катиона в системе никогда не бывает высокой (как, впрочем, и в случае с атомарным бромом), но она вполне достаточна, чтобы эффективно вести основную реакцию.
Атомы водорода метальной группы довольно инертны по отношению к атаке заряженных частиц. Напротив, поляризуемая система я-электронов ароматического ядра легко взаимодействует с активными катионами, в частности с Вг+. В упрощенном виде это взаимодействие включает последовательность, по-казанную на схеме 2.5. На первой стадии реакции одна из электронных пар ароматической системы сдвигается к атакующему катиону, завязывая связь С—Вг, что сопровождается перераспределением электронной плотности с возникновением положительного заряда на атоме, несущем метальную группу, как это показано в структуре а-комплекса 13. Отщепление протона из этого комплекса и восстановление ароматической системы связей дает продукт 8*.
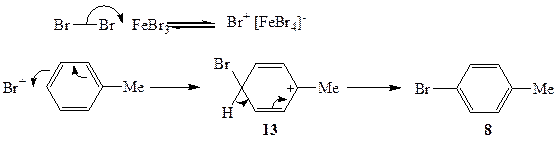 Схема 2.5
Схема 2.5
|
Показанные схемы механизмов, несмотря на их крайнюю упрощенность, позволяют, тем не менее, четко сформулировать условия, которые должны соблюдаться, если мы намерены получать при бромировании толуола избирательно либо бензилбромид (7), либо я-бромтолуол (8). В первом случае реакцию надо проводить при облучении, и реагенты не должны содержать примесей, способных вызвать образование бром-катиона (например, нельзя использовать без очистки толуол, хранившийся в железных бочках и содержащий следы ржавчины). Что касается ионного бромирования, приводящего к и-бромтолуолу (8), го его лучше проводить в темноте, иначе почти неизбежно побочное образование продукта радикального замещения, бензилбромида (7).
Многие другие реакции в ароматическом ряду протекают по механизму, показанному выше для образования соединения 8 (электрофильное замещение). К их числу, в частности относится реакция толуола с ацетилхлоридом в присутствии хлорида алюминия [реакция (3) ?ia схеме 2.3 (реакция Фриде-ля—Крафтса)]. Здесь реагентом является комплекс CH3COCI-AICI3, который служит источником катиона ацетилия, СН3СО+, реагирующего с толуолом с образованием а-комплекса, аналогичного комплексу \Ъ.
Сопоставим теперь другую пару реакций, итогом каждой из которых является присоединение водорода к толуолу [(реакции (4) и (5), схема 2.3]. Исчерпывающее гидрирование 6, приводящее к метилциклогексану (10), требует использования катализаторов, содержащих металлы VIII группы. Этот процесс протекает в адсорбционном слое на поверхности катализатора по довольно сложному механизму, суть которого сводится к активации водорода и субстрата, причем водород в этом состоянии может быть в первом при-бдижении описан как атомарный. В таком состоянии водород достаточно легко может присоединяться по двойным связям субстрата, адсорбированного на том же катализаторе, что в случае толуола и приводит в конце концов ^соединению 10. Очевидно, что для успешного проведения такого восста-давления необходимо наличие катализатора с хорошо развитой поверхностью, высокая концентрация водорода (повышенное давление) и отсутствие ^субстрате примесей, способных эффективно блокировать активные центры катализатора (каталитические яды).
к. Реакция (5) известна как реакция Берча. Она проводится с помощью металлов I группы (обычно натрия) в жидком аммиаке в присутствии донора протонов (обычно спирта). Ее результатом также является присоединение водорода, но в этом случае не трех, а лишь одной молекулы с образованием циклогексадиена (11). Причина подобного хода реакции становится ясной, если рассмотреть механизм процесса.
Растворение щелочного металла, например натрия, в жидком аммиаке — это в сущности электролитическая диссоциация, непосредственным результатом которой является образование системы, содержащей ионную пару: оольватированный катион металла — сольватированный электрон (схема 2.6). Первой стадией реакции Берча является атака сольватированного эдектрона на ароматическое ядро с образованием анион-радикала 14. Последний отрывает протон от спирта, давая при этом радикал 15. Этот радикал Присоединяет второй электрон, давая карбанион 16, взаимодействие которо-toco второй молекулой донора протона дает диен 11.
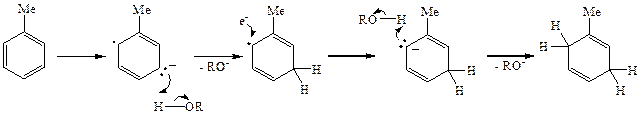 Схема 2.6
Схема 2.6
|
Образование именно диена 11, а не других изомеров определяется тем, что из всех возможных альтернативных структур для первично образующегося анион-радикала наиболее стабильной является структура 14.
Итак, управление степенью восстановления толуола может быть обеспечено достаточно просто и надежно за счет выбора путей проведения этой реакции — каталитическим гидрированием или восстановлением по Берчу.
Еще одна из показанных реакций толуола — окисление перманганатом с образованием бензойной кислоты (12) [(реакция (6), схема 2.3] — также является радикальным процессом, до некоторой степени аналогичным броми-рованию. Она состоит в последовательном замещении всех трех атомов водорода метальной группы с образованием карбоксильной группы. Интересно, что направленность этой реакции мало чувствительна к вариациям условий ее проведения. Это окисление может быть выполнено в воде или в среде органического растворителя, при комнатной температуре или нагревании — продуктом всегда будет бензойная кислота, хотя ее образование будет протекать с разной скоростью.
Показанные на схеме 2.3 реакции толуола иллюстрируют еще одну из особенностей большинства органических реакций, а именно тот факт, что в них принимают участие высокоактивные промежуточные продукты (интермедианты), строение и свойства которых определяют как направленность реакции, так и саму возможность ее осуществления. В наших примерах это были бром-катион и атомарный бром, сг-комплекс 13, анион-радикал 14 и карбанион 16. Однако интермедиаты ни в коем случае нельзя отождествлять с переходными состояниями, хотя нередко по строению и энергетике они могут быть близки последним. Различия между ними можно проиллюстрировать энергетическим профилем реакции, представленном на рис. 2.4.
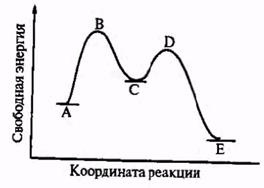 Рис. 2.4
Энергетический профиль реакции, включающей образование интермедиата. А – начальная система, В и D - переходные состояния; С - интермедиат; Е – конечный продукт.
Рис. 2.4
Энергетический профиль реакции, включающей образование интермедиата. А – начальная система, В и D - переходные состояния; С - интермедиат; Е – конечный продукт.
|
Интермедиат С — это вполне определенное химическое соединение, которое в принципе может быть выделено и охарактеризовано. Его отличие от «нормальных» соединений А и Е — чисто количественное, связанное с большим запасом энергии и с соответственно более низкими барьерами, отделяющими его от стабильных продуктов. Именно эти особенности и определяют высокую реакционную способность интермедиатов: благодаря большому за-досу энергии их реакции даже с малоактивными веществами оказываются лермодинамичски разрешенными, а благодаря низким барьерам эти реакции -могут протекать достаточно быстро.
Переходное состояние — это нечто совсем иное. Это именно мгновенное даястояние реагирующей системы в ее динамике, которое в принципе не может существовать в статике. Энергетический профиль показывает это на своем языке: отсутствие потенциальных барьеров для В и D делает невозможным даже их кратковременное существование. Система, достигшая максимума В или D, должна немедленно скатиться до состояния с более низкой энергией (А, С или Е), где она может закрепиться благодаря наличию потенциальных барьеров. Рис. 2.4 иллюстрирует еще одну важную черту этих реакций. Барьер между исходными соединениями и интермедиатами выше, чем между интерме-арнятами и конечными продуктами. Это означает, что именно образование интермедиата С будет самой медленной, лимитирующей, стадией процесса, показанного на этой схеме. Превращение же интермедиата С → Е будет быстрой стадией, поскольку система уже поднята почти до уровня потенциального барьера этой стадии.
Из сказанного можно вывести два заключения. Во-первых, интермедиа--1Ы могут существовать в типичной реакционной системе лишь в очень низ-f*HX концентрациях, поскольку они расходуются быстрее, чем накапливают--cgv Поэтому их выделение в индивидуальном состоянии или просто регистрация инструментальными методами могут оказаться достаточно сложной -задачей. Во-вторых, если удается каким-либо образом получить интермеди-far в виде стабильного соединения, то его реакции с субстратами могут про-гводиться в гораздо более мягких условиях и с большей селективностью. Это утверждение требует некоторых пояснений.
Поскольку интермедиаты, как мы уже отмечали, являются высокоактивными веществами, то их образование требует значительной затраты энергии, ст.е. применения высокоактивных реагентов, высокой температуры, сильных -кислот или оснований. Если проводить стадию генерации этих интермедиатов в присутствии субстратов, то может оказаться, что в применяемых жест--кнм условиях эти субстраты будут не только реагировать с образующимися «нтермедиатами, но и претерпевать совсем иные, побочные превращения, 'несвязанные с основной реакцией. Этих осложнений можно избежать, если удается разработать метод генерации интермедиата как стабильного соединения с тем, чтобы далее вводить его в реакцию с требуемым субстратом. При таком постадийном проведении процесса обеспечивается возможность проведения каждой из его стадий (генерации интермедиата и его реакции с субстратом) в оптимальных для нее условиях. Преимущества подобного подхода очевидны, и поэтому неудивительна наблюдаемая тенденция как можно шире использовать его при проведении самых разных превращений. Хотя мы еще неоднократно будем возвращаться к обсуждению этого важного момента, уместно здесь рассмотреть один из поучительных примеров.
Ацетилированиетолуола по Фриделю—Крафтсу [(реакция (3), схема 2.3)] проходит удовлетворительно при комнатной температуре (типичный выход кетона 9 — 70%), но ароматические системы, содержащие электроноакцеп-торные заместители, например галоген- или нитрозамещенные арены, реагируют весьма вяло, а форсирование реакции нагреванием может во многих случаях приводить к нежелательным осложнениям. Роль катализатора в этой реакции состоит в поляризации связи МеСО–Сl, что в пределе огвечает образованию формального интермедиата — соли ацетилия МеСО+АlСl4-. Оказалось, что на самом деле подобного рода соли могут быть довольно легко получены как стабильные (в растворе или в твердом состоянии) соединения, например, MeCO+SbF6-. Поскольку ацилий-катион уже присутствует «в готовом виде» в такого рода реагентах, то с их участием реакции Фриделя— Крафтса могут проводиться при пониженных температурах даже для малоактивных ароматических субстратов.
Итак, на ряде примеров мы видели, что существуют достаточно широкие возможности для того, чтобы управлять реакционной способностью органических соединений и направлять их превращения в желаемую сторону путем тщательного выбора необходимых для этого реагентов и условий, оптимальных для протекания требуемой реакции. Разнообразие органических реакций поистине поразительно, однако далеко не все они могут служить эффективными инструментами в направленном органическом синтезе. Действительно, основные пути взаимного превращения органических соединений уже были найдены к 30-м годам XX в., и уже не существовало принципиальных препятствий для реализации превращения «чего угодно во что угодно» или, иначе говоря, для синтеза соединений любой сложности. Иными словами, уже в те времена органическая химия могла решать, по крайней мере в принципе,синтетические задачи любой сложности. Однако потребовалось еще несколько десятилетий для того, чтобы теоретически возможное превратилось в практически реализуемое*. Такая кардинальная трансформация самого «образа» органического синтеза стала возможной в первую очередь благодаря действительно революционным достижениям в области создания новых синтетических методов. Конечно, многие из этихметодов были созданы благодаря открытию новых реакций, но не меньшее значение имела разработка проблем, связанных с синтетическим использованием уже хорошо известных реакций, так сказать, возведением этих реакций в ранг синтетических методов. Посмотрим, что же для этого требуется от органической реакции.
Дата добавления: 2015-04-05; просмотров: 2106;