Синтез как инструмент исследования
Во всех обсуждавшихся выше примерах синтез выполняет чисто препаративную функцию, т.е. поставляет нужные вещества. В принципе для решения таких задач не имеет значения, каким именно путем было получено данное вещество: его можно было выделить из природных источников или получить микробиологическим путем (хотя в большинстве случаев именно химический синтез оказывается наиболее общим и надежным способом).
Однако в некоторых областях синтез используется не просто в качестве удобного «подручного средства», а составляет самую суть задачи. Речь идет, в первую очередь, о встречном синтезе природных соединений или соединений, впервые полученных в результате неизвестных ранее химических превращений. В таких случаях наиболее надежным и бесспорным доказательством справедливости структуры, выведенной на основании обычных методов анализа, является его химический синтез и установление идентичности полученного вещества заведомого строения с исследуемым веществом. Подобное утверждение сегодня может показаться устаревшим. Действительно, такие мощные современные методы структурного анализа, как спектроскопия в ультрафиолетовой, видимой и инфракрасной областях, ядерный магнитный резонанс, масс-спектрометрия высокого разрешения, казалось бы, обеспечивают возможность быстрого установления структур любой сложности. Однако нельзя забывать, что трактовка результатов, получаемых любым из этих физических методов, базируется прежде всего на аналогиях с известными соединениями и потому тем более надежна, чем ближе структура изучаемого соединения к уже изученным. В случае же необычных новых типов структур интерпретация спектров в структурных терминах может стать далеко не тривиальной задачей. Конечно, существует метод, свободный от этих ограничений, а именно рентгеноструктурный анализ (РСА). Этот метод справедливо считается абсолютным методом установления структуры, однако ему присущи ограничения другого, технического характера — он применим лишь в тех случаях, когда можно получить монокристалл исследуемого вещества, что достижимо далеко не всегда.
Другой, классический подход к установлению структуры вещества основан на его деструкции, т.е. на химической «разборке» молекулы на составные части и логической реконструкции исходной структуры на основе информации о строении этих составных частей. Этот путь, несмотря на его универсальную применимость, очень трудоемок и предполагает наличие значительных количеств анализируемого вещества. К тому же в ходе подобной деструкции почти неизбежна потеря части информации (в особенности сте-реохимической) относительно строения тех участков молекулы, по которым производится ее деструкция.
От всех упомянутых выше ограничений свободен встречный синтез. Если структура исследуемого вещества подтверждена его встречным синтезом, то она может считаться полностью доказанной, и этот «вердикт» не подлежит пересмотру.
Нередки ситуации, когда встречный синтез вообще оказывается единственным средством выбора между несколькими альтернативными структурами изучаемого вещества. Так бывает в тех случаях, когда вещество доступно в ничтожно малых количествах — в долях миллиграмма или даже микрограммах, — которых явно мало для использования деструктивных методов анализа и даже Для применения современных спектральных методов. В то же время этих количеств вполне может хватить для идентификации вещества, т.е. установления тождественности двух его образцов, например, синтетического и природного.
 Схема 1.16
Схема 1.16
|
Именно встречный синтез позволил окончательно установить структуры таких природных веществ, как уже упоминавшиеся ювенильный гормон (8) или бомбикол (12). Ниже мы рассмотрим еще несколько более недавних примеров.
В 1976-78 гг. ценой напряженного труда удалось выделить в чистом виде один из активнейших половых феромонов самки таракана Periplaneta ameriсапа.Активность выделенного вещества, перипланона В, оказалась поистине удивительной — порог чувствительности особи составлял всего 10-13 г, и оно рассматривалось как перспективное средство борьбы с этим вездесущим насекомым. Однако в распоряжении исследователей имелось лишь ничтожно малое количество этого вещества (около 200 мкг перипланона В, выделенные из 75000 особей). Даже использование всего арсенала средств инструментального анализа не позволило однозначно установить его строение. С помощью этих методов удалось лишь выяснить некоторые основные особенности структуры, а именно: строение углеродного скелета, характер и распределение функциональных групп, как это показано в формуле 59а (схема 1.16), но вопрос о стереохимии молекулы оставался открытым. Эту самую важную вй6жную часть задачи удалось решить лишь после того, как был выполнен полный синтез трех из четырех возможных диастереомеров перипланона В |2ба]. Прямое сравнение синтезированных образцов с природным позволили установить, что стереохимия последнего соответствует показанной в формуле 59б. Минорный компонент природного феромона, перигшанон А, был в еще меньших количествах, и по данным первоначальных спектральных исследований ему была ошибочно приписана структура 60а.Истинную структуру 60Ь удалось выяснить только путем полного синтеза этого соединения [26Ь]. Благодаря интенсивным синтетическим исследованиям в этой области, был получен также ряд биологически активных аналогов 59а (как, например, 61) из достаточно доступных предшественников [26с].
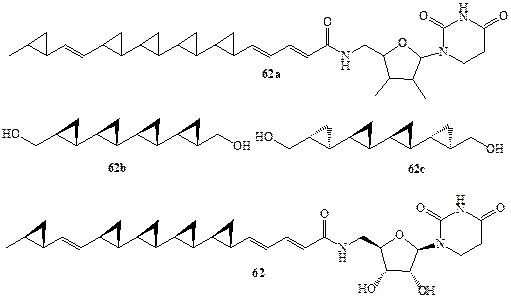 Схема 1.17
Схема 1.17
|
В 1990 г. японским химикам удалось выделить из культуральной жидкости микроорганизма Slreptoverticillium fervens новый антигрибковый агент обозначенный как FR-900848, для которого была установлена общая структура 62а (схема 1.17). Весьма интересной оказалась активность этого агента против нитевых грибов, особенно опасной инфекции для больных СПИДом или диабетом [27]. В течение ряда лет стереохимию этого соединения не удавалось установить, так как даже самые изощренные методики ЯМР-спектроскопии оказались бессильными для решения задачи установления стереохимии для полициклопропанового остова 62а, содержащего 10 хиральных центров. Было также
очевидно, что в этом случае вряд ли разумно было пытаться решить эту проблему путем синтеза всех (210!) диастереомеров этого соединения. Однако рассмотрение возможных путей биосинтеза этого соединения в живой клетке позволило предположить, что наиболее вероятной является полностью транс-конфигурация (all-trans) заместителей во всех циклопропановых остатках [27а]. При частичной деградации природного продукта было получено симметричное производное кватерциклопропана, которому можно было приписать структуру 62Ь или 62с. Оба этих диастереомера были получены независимым синтезом с использованием стереоспецифичных реакций, что позволило однозначно установить полностью транс-, полностью сан-конфигурацию (all-trans, all-sin) продукта деградации, т.е. структуру 62Ь. Этот результат в сочетании с рядом других данных позволил установить полную структуру FR-900848, показанную в формуле 62 [27Ь]. Попутно заметим, что вскоре был выделен следующий представитель полициклопропанов из культуральной жидкости ряда видов стрептомицетов Streptomyces spp. Это соединение, проявляющее значительную активность как ингибитор отложения липидов на стенках артерий, отличается от соединения 62 наличием еще одного циклопропанового звена и, по-видимому, принадлежит к тому же стереохимическому ряду [27с].
Дополнительные сложности при установлении строения могут возникать из-за повышенной лабильности некоторых природных соединений. В 1969 г. в результате тщательных биомедицинских исследований было обнаружено, что в тромбоцитах продуцируется, хотя и в очень малых количествах, вещество, являющееся чрезвычайно мощным фактором сужения сосудов и агрегации тромбоцитов. Имелись довольно серьезные основания полагать, что результатом «перепроизводства» этого фактора, тромбоксана А2 (ТхА2), может быть сердечный приступ или инсульт. Выделить этот фактор удалось лишь в 1975 г., но его крайняя нестабильность (период полураспада этого вещества в водном растворе при 37°С составляет всего 34 с!), если и не исключала, то, по крайней мере, крайне ограничивала возможность использования для установления его строения стандартных физико-химических методов. Однако не составило большого труда выяснить, что образующееся в результате разложения тромбоксана биологически инертное вещество имеет строение 63а (схема 1.18) [28а]. Этот факт в сочетании с данными, полученными при изучении путей биогенеза всего семейства простаноидов, позволил предложить структуру 63как наиболее вероятную для тромбоксана А2. Эта структура казалась настолько необычной для природного соединения, что на протяжении почти 10 лет она неоднократно оспаривалась. Тем не менее, именно данная структура была однозначно подтверждена в 1985 г., когда в лаборатории Стилла [28Ь] был выполнен полный синтез ТхА2. Интересно, что в данном случае только один единственный критерий мог быть использован для доказательства идентичности образцов природного и синтезированного веществ, а именно тождественность их биологической активности в серии стандартных тестов. Исключительная нестабильность соединения 63 оказалась также серьезным препятствием для экспериментального изучения его физиологических функций, что побудило предпринять серию исследований, направленных на синтез химически стабильных аналогов 63, Среди множества последних одним из наиболее эффективных заменителей 63 оказачось соединение 64 [28с].
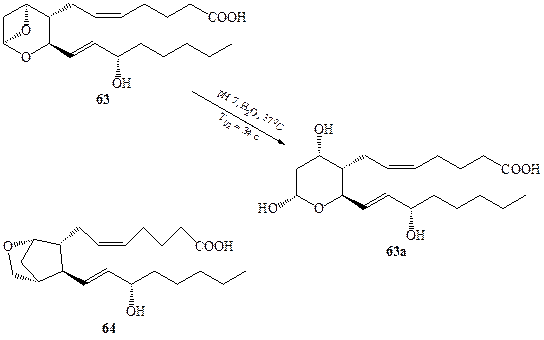 Схема 1.18
Схема 1.18
|
Однако помимо той, иногда действительно ключевой роли, которую играет синтез при решении задач установления строения, у него есть еще другая, менее очевидная, но более общая и, пожалуй, более важная функция. Дело в том, что синтетическое исследование само по себе является наиболее мощным инструментом активного познания химии синтезируемых соединений. Действительно, только глубокое понимание химического своеобразия -Органического соединения позволяет успешно осуществить его целенаправленный синтез, но сами знания, необходимые для подобного понимания, эффективно накапливаются именно в ходе выполнения этого синтеза. Почему это так?
Первоначальный план синтеза обычно строится на хорошо известных синтетических методах, принципиальная пригодность которых для решения конкретной задачи не вызывает особых сомнений. И если все идет по плану, то мы тем самым получаем экспериментальное подтверждение справедливости наших предсказаний о химии конкретных соединений, участвующих в предпринятом синтезе. По этому поводу один из величайших синтетиков XX века лауреат Нобелевской премии Р. Вудворд писал: «Вряд ли можно отрицать, что успешный исход синтеза, состоящего из более чем 30 стадий, является суровым испытанием способности науки к предвидению, а также проверкой ее познавательной мощи в сфере изучаемых объектов» [29].
Однако нередко при вторжении в новую «сферу изучаемых объектов» хорошо апробированные методы не срабатывают, и здесь и начинается самое интересное. Во-первых, сам факт такой «осечки» — это уже небольшое (а иногда и значительное) открытие — обнаружение неожиданной химической особенности, присущей изучаемой структуре. Причем маловероятно, чтобы эта особенность была обнаружена «просто» при изучении химии данного соединения вне связи с его синтезом. Маловероятно именно потому, что речь идет о хорошо изученных реакциях, результат которых казался точно предсказуемым и поэтому, если бы не потребности синтеза, то вряд ли имело смысл ставить такой эксперимент. Во-вторых, существование конкретной синтетической цели не позволяет исследователю ограничиться индифферентной констатацией факта, что такая-то реакция в данном случае странным образом не идет или идет, но не так, как предполагалось. Затруднениенужно преодолеть, и наиболее эффективный путь для этого — понять причину возникновения аномалий, т. е. глубже разобраться в химии изучаемых соединений. Понимание причин наблюдаемых осложнений может помочь в разработке нового варианта примененного метода. Если и таким путем преодолеть препятствие не удается, то приходится искать какие-то новые пути и привлекать для этого и методы и идеи из других, часто отдаленных областей химии. Такой напряженный и многогранный целенаправленный поиск решения дает в качестве «побочного продукта» новые и углубленные знания о реакционной способности органических соединений — знания, которые вряд ли бы были получены в обозримое время, если бы не возникло проблем с решением поставленной синтетической задачи.
В справедливости сказанного читатель легко может убедиться, обратившись к серии блестящих синтезов, выполненных в группе Кори. В своей Нобелевской лекции Кори специально подчеркнул, что «ключом к успеху множества многостадийных синтезов, которые были осуществлены в нашей лаборатории за последние годы, было изобретение новой методологии» [30], Согласно его оценкам, не менее 50 новых методов было создано в ходе этих исследований.
Более того, подчас именно благодаря «осечкам» при применении хорошо известных методов к решению конкретной и казалось бы частной синтетической задачи удавалось обнаружить принципиально новые особенности строения и реакционной способности исследуемых объектов. Их последующее изучение приводило к результатам, важным для всей органической химии, далеко за пределами «сферы изучаемых объектов». Поясним это утверждение некоторыми примерами.
В 1900 г. Гомбергом проводилось исследование, целью которого был синтез гексафенилэтана (65). По тем временам эта структура казалась довольно экзотичной, но на самомделе тем же Гомбергом ранее (в 1897 г.) уже был синтезирован, хотя и с незначительным выходом, тетрафенилметан, и, вообще говоря, не было оснований ожидать особых осложнений при синтезе 65, «всего лишь» следующего представителя перфенилированных производных алканов. Однако использование стандартного в то время метода создания углерод-углеродной связи — конденсации галогенпроизводных в присутствии металлов(схема 1.19) — привело к более чем странным результатам: вообще не наблюдалось образования углеводорода 65 при реакции трифенилхлорметана с каким-либо из металлом (Zn, Ag или Hg) в обычных условиях проведения подобного рода конденсаций. При этом в качестве единственного продукта реакции был получен пероксид 65а [31а]. Естественно, что следующим шагом было проведение реакции в условиях, исключающих наличие кислорода воздуха (в атмосфере СО2), и в этом случае удалось получить углеводород, отвечающий по составу гексафенилэтану (65). Однако совершенно неожиданно было обнаружено, что полученный углеводород ведет себя в растворе как димер, способный к диссоциации в растворе на две идентичные субъединицы, и вприсутствии кислорода он быстро превращается в тот же пероксид 65а. Анализ этих и других экспериментальных данных привел Гомберга к выводу о том, что на самом деле непосредственным результатом реакции трифенилхлорметана с металлом является образование трифенилметила как стабильной частицы 66а [31а,B], которая в растворе может претерпевать обратимую димеризацию в гексафенилэтан (65). В то время требовалась немалая смелость для подобного заявления, подразумевавшего возможность существования соединений, в которых углерод трехвалентен, поскольку господствовало убеждение, что углерод бывает либо четырехвалентным, либо — в редких случаях — двухвалентным (например, в монооксиде углерода или изонитрилах, RN=C). Поэтому вполне естественно, что результаты синтетических опытов Гомберга были встречены с большим сомнением.
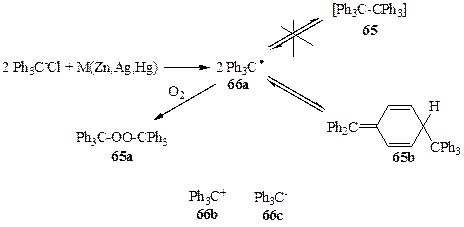 Схема 1.19
Схема 1.19
|
Вскоре, однако, достоверность этих результатов была строго подтверждена, и стало ясно, что итогом «синтетической неудачи» Гомберга* явилось открытие свободных радикалов — нового класса химических частиц, производных трехвалентного углерода. Дальнейшие исследования показали, что для такой же трифенилметильной системы могут быть получены и производные трехвалентного углерода других типов, а именно соли трифенилметил-катиона (66Ь) и трифенилметил-аниона (66с).
Получение бба-с, первых представителей соединений нового структурного типа как стабильных веществ, а также выяснение факторов, обусловливающих их стабильность и особенности реакционной способности, послужило ивщным импульсом для разработки новых представлений о механизмах органических реакций, которые предусматривали промежуточное образование подобного рода интермедиатов. Возникновение этих представлений сыграло ключевую роль в становлении классической теории органической химии. Подчеркнем еще раз, что первичным толчком, «запустившим» всю серию столь важных по последствиям исследований, явилась работа Гомберга, преследовавшая сугубо препаративные цели.
Не менее выразительный пример первостепенной роли синтеза в развитиитеоретических представлений может быть найден в истории полного и частичного синтеза стероидных гормонов и их аналогов. Исследователи, работавшие в этой области в 1930—40-х годах, встретились с рядом неожиданных проблем как при построении углеродного скелета, так и при осуществлении некоторых иногда вполне тривиальных превращений, таких, как присоединение по связи С=С или С=О, раскрытие оксиранового цикла или даже превращение спиртов в соответствующие галогенопроизводные. Потребности синтеза не только заставили химиков разработать альтернативные методы, позволявшие осуществлять такие превращения, но и побудили обратиться к изучению причин наблюдаемых аномалий. Именно благодаря глубокому анализу особенностей реакционной способности функциональных групп в конформационно закрепленных системах (а к таким системам относится тетрациклический остов стероидов) и удалось сформулировать основные понятия современного конформационного анализа. Напомним, что еще в 1890 г. Заксе [32а] предположил, что циклогексан не является плоской молекулой и сделал вывод о том, что «все монозамещенные производные циклогексана могут существовать по крайней мере в виде двух модификаций». Поскольку в то время не имелось никаких экспериментальных данных в пользу этого, вообще говоря, вполне разумного предположения (вспомним, хотя бы тот факт, что к этому моменту тетраэдрическая модель атома углерода Вант-Гоффа и Ле Беля уже была общепринятой), о нем никто особенно и не вспоминал в последующие 60 лет, хотя за этовремя появился ряд теоретических и физико-химических исследований, свидетельствовавших о правомерности подобного рассмотрения. Но в полной мере прозорливость предположения Заксе могла быть оценена лишь после публикации в 1950 г в журнале Experientia короткого сообщения под названием «Conformation of the Steroid Nucleus» [32b]. Автор этой работы Бартон проделал огромную работу по сбору и обобщению многочисленных и противоречивых данных по реакционной способности различных замешенных стероидов и собрал убедительные доказательства того, что наблюдаемые различия обусловлены прежде всего значительным различием свойств заместителей в зависимости от того, находятся они в аксиальном или экваториальном положениях, а также особенностями стереохимии всей молекулы. На основе этих представлений вскоре была развита вся концепция конформационного анализа [32с], и пионерский вклад Бартона в ее создание был справедливо отмечен Нобелевской премией 1969 г.
В настоящее время представления конформационного анализа стали неотъемлемой частью теоретических основ органической химии. Трудно представить себе, что на протяжении почти 100 лет этой концепции вообще не существовало и что ее рождение было обусловлено более всего неотложными потребностями органического синтеза.
Органический синтез сегодняшнего дня успешно решает задачи получения молекулярных конструкций невероятной сложности. Закономерно, что на каждом этапе усложнения целей органического синтеза возникают неожиданные синтетические проблемы, обнаруживаются специфические особенности строения и реакционной способности, что не только стимулирует разработку новых синтетических методов, но и служит постоянным источником фактического материала для углубления уже сложившихся и создания новых концепций теоретической органической химии. Поэтому легко представить, насколько беднее и приземленнее выглядела бы органическая химия, если бы по какой-либо причине ей пришлось отказаться от синтеза как одной из главных своих задач*.
«Химия создает свой предмет...»
Еще в 1860 г. выдающийся химик XIX в. М. Бертло писал: «Химия создает свой предмет. Эта творческая способность, подобная искусству, коренным образом отличает химию от остальных естественных и гуманитарных наук» [33]. Попробуем разобраться, на чем основывалось подобное представление об исключительном положении химии в ряду других наук.
Действительно, во все времена естествознание занималось изучением Природы, поисками внутренней связи явлений и законов, управляющих этими явлениями. Природа для ученого всегда являлась изначальной данностью, которую надо было исследовать. Так, биолог изучает живую природу в том виде, в каком она сформировалась в условиях Земли. Астроном изучает уже существующие планеты, звезды, галактики и, наконец, всю Вселенную как целое. Объект исследования химика-органика — органические соединения, их свойства, реакции и закономерности поведения. Однако в отличие от своих коллег-естественников химик должен был сначала создать свой объект исследования, причем создать в самом прямом и точном смысле слова, т.е. синтезировать вещества, которые в Природе (или по крайней мере на Земле) тогда не существовали. В этом смысле органическая химия действительно принципиально отличается от всех других естественных наук, и с самого своего начала она составляет систему, которая черпает в самой себе как объекты исследования, так и проблемы, требующие решения, и развивается по своим вутренним законам. Аналогию такой способности к саморазвитию можно найти разве что в математике.
Так обстояло дело во времена Бертло, и в значительной мере так оно обстоит и сейчас, хотя исключительность органической химии в смысле создания своего объекта исследования несколько поколебалась с появлением совсем новых областей науки, таких, как физика твердого тела, нелинейная оптика, генная инженерия и т.д., развитие которых в значительной мере основано на создании сложных искусственных объектов.
Тем не менее основная мысль Бертло остается справедливой и сегодня: органическая химия выступает в роли подлинного творца, постоянно создавая ту самую искусственную природу, которую сама же и исследует, развивает и находит ей области применения.
Более того, и это особенно интересно, свойства этой искусственной природы оказываются столь же разнообразными, неожиданными и неисчерпаемыми, как свойства «обычной» природы. Это и составляет принципиальное отличие синтетических органических соединений от других классов искусственных объектов. В самом деле механические, электрические или логические структуры, создаваемые человеком, могут быть беспрецедентно сложными, не имеющими прототипов в Природе. Но при всей их сложности никаких качественно новых свойств, которые не могли быть предположены на стадии Проекта, в них обнаружиться не может, поскольку они проектировались и создавались для совершенно определенных целей.
Например, если мы проектируем и строим самолет, то он может быть лишь хорошим или плохим самолетом, но ни при каких обстоятельствах не окажется вдруг магнитофоном или мясорубкой. Напротив, если мы синтезируем новое соединение, предназначенное служить лекарством, то, вообще говоря, нет никакой гарантии, что оно не окажется токсином, дефолиантом, фотосенсибилизатором или еще чем-то совершенно непредвиденным. Столь же неожиданными могут оказаться результаты синтетических исследований, не преследующих каких-либо прикладных целей.
Так, в середине 1880-х годов молодой русский химик Зелинский, работавший в лаборатории Майера в Германии, разрабатывал новую схему получения тетрагидротиофена (67) из 2-хлорэтанола (68) с помощью подкупающе простой последовательности реакций, показанной на схеме 1.20. Однако осуществление синтеза пришлось внезапно остановить на стадии получения ключевого полупродукта, а именно b,b'-дихлордиэтилсульфида (69). Вместо того, чтобы попробовать осуществить последнюю стадию синтеза, внутримолекулярную циклизацию по реакции Вюрца (кстати, в настоящее время можно с уверенностью утверждать, что при обработке 69 металлом ничего, кроме элиминирования не могло произойти), Зелинскому пришлось провести несколько недель в больнице из-за серьезнейших ожогов, вызванных контактом с таким простым и вполне невинно выглядевшим (на бумаге!) соединением, которое позднее приобрело вполне заслуженную дурную славу под названием «иприт». Однако подобной «зловредной» физиологической активностью вовсе не исчерпываются свойства иприта, и его открытие принесло
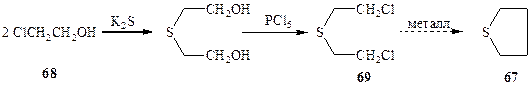 Схема 1.20
Схема 1.20
|
человечеству не только бедствия. Детальное исследование механизма его действия, вызванное суровой необходимостью в ходе первой мировой войны, привело к созданию нового и по тем временам наиболее эффективного направления в лечении злокачественных опухолей, основанного на использовании иприта и его структурных аналогов в качестве химиотерапевтических средств.
Рассмотренный пример (а число таких примеров исчисляется сотнями!) наглядно показывает, что органические соединения, созданные руками человека, в такой же мере могут служить источником совершенно неожиданных открытий, как и нерукотворные объекты исследований, поставляемых Природой.
Причины такого своеобразия органической химии лежат прежде всего в безграничности числа возможных органических соединений, а следовательно, в безграничном многообразии их свойств (частным проявлением этого многообразия является сам факт существования жизни на Земле).
Общеизвестно, что уникальность углерода состоит в сочетании двух свойств: его четырехвалентности и способности образовывать прочные связи как с другими атомами углерода, так и с атомами многих других элементов. Именно поэтому число возможных органических соединений оказывается бесконечно большим, в строгом смысле этого слова.
Как формулируется понятие бесконечности в математике, скажем, в простейшем случае бесконечности натурального ряда чисел? К любому сколь угодно большому числу можно прибавить единицу и получить следующий член этого ряда, с которым можно проделать ту же операцию, и т. д. Аналогично к любой сколь угодно сложной органической структуре можно присоединить (по крайней мере, теоретически), например, метальную группу и получить новое соединение. С той только разницей, что такую операцию со сложной органической молекулой можно выполнить множеством различных способов, а присоединять можно отнюдь не только метильную группу. Множественность вариантов усложнения проявляется на самых ранних этапах, начиная с молекул, содержащих всего несколько атомов, и число таких вариантов возрастает примерно пропорционально числу уже имеющжся в молекуле атомов углерода. В таком «ветвящемся дереве» множества структур коэффициент ветвления будет монотонно возрастать с ростом уже пройденных точек ветвления. Общее число членов такой системы должно расти по закону, близкому к факториалу (л!, где п — число атомов углерода в молекуле). Это означает, что даже в пределах не очень больших органических молекул число возможных структур становится поистине астрономическим.
Рассмотрим, например, соединения состава Qo (схема 1.21), относящиеся к узкому классу — насыщенным алифатическим кислотам обшей формулы 70, в которых заместителями R1 и R2 в любом возможном положении могут быть любые из десяти показанных на схеме групп.
Число подобных структур, возникающих просто при вариации природы и положения всего лишь одной из групп, R1 или R2, составит 1029. Если варьировать обе группы, то общее число возможных комбинаций составит 1029- Ю29, что примерно в 107 раз превышает число всех атомов Земли. Всего углерода, имеющегося в нашей Галактике, не хватит на то, чтобы получить все соединения из этого набора даже в миллиграммовых количествах. Каждый из третичных атомов углерода в соединениях 70 является асимметрическим центром, и поэтому любое из них может быть представлено 229 стерео-изомерами, что увеличивает общее число структур типа 70 примерно до 5,4 ■ 1066. Для их синтеза (по 1 мг каждого) не хватит уже всех нуклонов во всей наблюдаемой Вселенной. Так, от абстрактной математической бесконечности мы приходим к вполне реальному, поистине неисчерпаемому многообразию органических соединений.
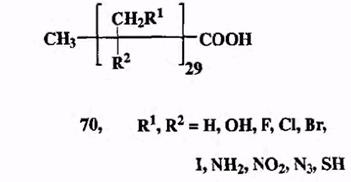 Схема 1.21
Схема 1.21
|
Каковы же источники всего этого многообразия? Как бледная схема теоретически возможных виртуальных структур расцвечивается полнокровными красками реально существующих веществ? Таких источников два: природный (ископаемое органическое сырье и современные живые организмы), и искусственный (органический синтез).
Органическая химия зародилась как химия соединений, выделяемых из живых организмов (чему она и обязана своим названием). Однако природные соединения несмотря на огромное разнообразие их структур заполняют систему органических соединений очень прихотливым и — с чисто органохимической точки зрения — случайным образом, поскольку пути биосинтеза определяются прежде всего биологической целесообразностью, а вовсе не потребностями химической систематики.
Если взять любую рациональную классификацию органических соединений, например, по функциональным группам, и заполнить ее только структурами природных соединений, то мы увидим очень странную картину: отдельные кластеры, густо усеянные разнообразными структурами, области, содержащие лишь отдельные точки, и, наконец, огромные пустые области. В такой системе, например, будут щедро представлены неразветвленные алифатические кислоты с четным числом атомов углерода, но будет мало разветвленных кислот или кислоте нечетным числом атомов углерода; будет множество очень причудливо устроенных циклических и полициклических систем, но почти не встретится их простейших представителей. Редкими и «случайными» структурами будут представлены такие важнейшие классы, как алкилгалогениды, тиолы и сульфиды, нитро- и диазосоединения. Удивительно, но будут отсутствовать даже такие тривиальные соединения, как формальдегид, хлороформ, диэтиловый эфир или тетрагидрофуран. Мы уже не говорим о том, что многае важнейшие классы органических соединений, такие, как, например, различные типы металлоорганических соединений или борорганические производные, вообще никак не представлены в списке природных веществ.
Совершенно ясно, что с таким материалом, каким разнообразным бы он ни был, органическую химию, как науку, создать было бы невозможно. Именно поэтому с первых же своих самостоятельных шагов химики-органики с поразительной смелостью пошли по пути создания своего объекта исследования, синтезируя тысячи и тысячи неизвестных Природе веществ и изучая их свойства и взаимопревращения. Огромные усилия нескольких поколений ученых были потрачены на то, чтобы создать прежде всего фундамент фактов для новой науки — органической химии — и определить проблемы, которыми она должна заниматься. Без этого не могло бы состояться создание грандиозной области науки и промышленности и в конечном счете новой, искусственной природы. Эта созданная руками человека природа не только обеспечивает нас почти всем необходимым для повседневной жизни, но и становится на наших глазах все более значимым биогеохимическим фактором глобального масштаба*.
Здесь, конечно, не место излагать историю органической химии, но стоит, хотя бы схематически, проследить те главные линии ее развития, в которых определяющую роль играл и продолжает играть органический синтез.
Дата добавления: 2015-04-05; просмотров: 1653;