Теоремы Хоэнберга-Кона
DFТ базируется на модели Томаса-Ферми, но обоснование для нее было подведено только в теоремах Хоэнберга-Кона. Эти теоремы - основа DFT метода. Первая теорема - теорема существования. Вторая теорема– вариационный принцип квантовой механики сформулированный для функционала плотности.
Теорема 1: Существует взаимно однозначное соответствие между плотностью основного состояния электронной подсистемы, находящемся во внешнем потенциале атомных ядер, и самим потенциалом ядер.
Теорема 2: Энергия электронной подсистемы, записанная как функционал электронной плотности, имеет минимум, равный энергии основного состояния.
Традиционные методы определения электронной структуры, в частности, метод HF и производные от него, описывают систему многоэлектронной волновой функцией. Основная цель теории функционала плотности — при описании электронной подсистемы заменить многоэлектронную волновую функцию электронной плотностью. Это ведет к существенному упрощению задачи, поскольку многоэлектронная волновая функция зависит от 3N переменных — по 3 пространственных координаты на каждый из N электронов, в то время как плотность — функция лишь трёх пространственных координат.
Как правило, метод DFT используется с формализмом Кона — Шэма, в рамках которого задача об описании нескольких взаимодействующих электронов в статическом внешнем поле (атомных ядер) сводится к более простой задаче о независимых электронах, которые движутся в некотором эффективном потенциале. Этот эффективный потенциал включает в себя статический потенциал атомных ядер, а также учитывает кулоновские эффекты, в частности, обменное взаимодействие и электронную корреляцию.
Описание двух последних взаимодействий и представляет собой основную сложность метода теории функционала плотности в формулировке Кона — Шэма.
Простейшим приближением здесь является приближение локальной плотности, основанное на точном расчёте обменной энергии для пространственно однородного электронного газа, который может быть выполнен в рамках модели Томаса — Ферми и из которого можно получить также и корреляционную энергию электронного газа. В ряде случаев использование простого приближения локальной плотности дает результаты, соответствующие экспериментальным данным, причём вычислительная сложность метода невысока относительно других подходов к проблеме многих частиц в квантовой механике. В тоже время, метод был недостаточно точен для расчётов в области квантовой химии, пока в 1990-х годах не произошёл сдвиг в описании обменного и корреляционного взаимодействий.
В настоящее время DFT метод является одним из основных подходов в квантовой химии. Но, все ещё имеются проблемы в приложении метода к описанию межмолекулярных сил, в особенности ван-дер-ваальсовых сил и дисперсионного взаимодействия, а также в расчётах ширины запрещённой зоны в полупроводниках.
Сложности с учетом дисперсионного взаимодействия в рамках DFT (когда этот метод не дополняется другими) делают метод теории функционала плотности малопригодным для систем, где дисперсионные силы являются преобладающими (взаимодействия между атомами благородных газов) или сопоставимыми с другими взаимодействиями (в ряде органических структур). Решение этой проблемы является предметом современных исследований.
Рассмотрим уравнение Шредингера для системы электронов:
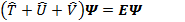
где оператор кинетической энергии электронов:
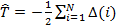
оператор, описывающий энергию взаимодействия электронов:
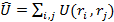
а оператор потенциальной энергии, описывающий взаимодействие электронов с ядрами:
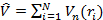
Очевидно, что первые два оператора универсальны для всех систем и каждая конкретная система отличается от остальных только оператором 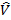 .
.
Фактически, теорема Хоэнберга—Кона означает, что выражение может быть обращено, то есть по заданной плотности частиц в основном состоянии, можно найти соответствующую волновую функцию основного состояния. Можно записать, что:
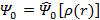 ,
,
Благодаря этому можно утверждать, что все наблюдаемые физические величины также являются функционалами. Если 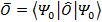
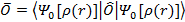 .
.
Тогда, согласно теореме Хоэнберга—Кона, для основного состояния электронной системы, находящейся в ядерном потенциале 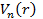 существует универсальный функционал
существует универсальный функционал 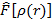 , не зависящий от системы, действующий на электронную плотность, который определяет полную энергию системы как
, не зависящий от системы, действующий на электронную плотность, который определяет полную энергию системы как
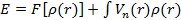 .
.
Используя это выражение, можно было бы найти 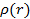 , используя вариационный принцип. В тоже время, так как форма функционала
, используя вариационный принцип. В тоже время, так как форма функционала 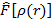 точно не известна, необходимы дальнейшие предположения.
точно не известна, необходимы дальнейшие предположения.
6-й учебный вопрос: Приближение локальной электронной плотности(LDA)
Наиболее часто используемой аппроксимацией является приближение локальной электронной плотности (local density approximation, LDA). Впервые оно было сформулировано Коном и Шэмом в 1965 году. В этом приближении функционал записывается в виде:
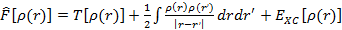
где 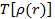 — электронная кинетическая энергия, а
— электронная кинетическая энергия, а 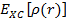 — функционал обменно-корреляционной энергии, выражающийся как:
— функционал обменно-корреляционной энергии, выражающийся как:
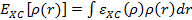 ,
,
где 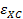 — обменная и корреляционная энергия на одну частицу электронного газа. Именно потому, что в этом приближении обменно-корреляционная энергия зависит от локальной плотности заряда в точке, приближение носит такое название.
— обменная и корреляционная энергия на одну частицу электронного газа. Именно потому, что в этом приближении обменно-корреляционная энергия зависит от локальной плотности заряда в точке, приближение носит такое название.
Корреляционный функционал вообще говоря является суммой корреляционного и обменного вкладов:
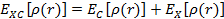 .
.
В LDA приближении обмен вычисляется как для однородного газа с плотностью, равной плотности в точке (LDA подходит для медленно меняющихся плотностей):
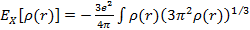 .
.
Применяя к (3.17) вариационный принцип и учитывая, что электронная плотность 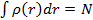 нормирована на число частиц, получим уравнение:
нормирована на число частиц, получим уравнение:
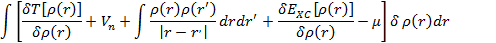
где m — множитель Лагранжа. Можно определить электронную
плотность с помощью волновых функций уровней:
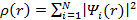 , (3.23)
, (3.23)
тогда кинетическую энергию можно определить следующим образом:
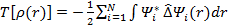
Тогда решение уравнения (3.22) является решением одноэлектронного уравнения Шредингера:
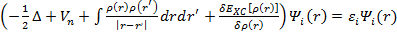 .
.
Эти уравнения называются уравнениями Кона-Шэма. Соответственно их решения являются одноэлектронными волновыми функциями, а собственные значения — энергиями соответствующих состояний.
Уравнение вообще говоря, нелинейно, так как оператор Гамильтона сам зависит от решения, поэтому для получения решения необходимо применять самосогласованный итерационный метод.
Стоит отметить, что 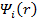 не являются спин-орбиталями в приближении HF, поэтому детерминант Слэтера, составленный из них, не будет волновой функцией системы в приближении HF. Несмотря на это, плотность заряда, выражаемая через
не являются спин-орбиталями в приближении HF, поэтому детерминант Слэтера, составленный из них, не будет волновой функцией системы в приближении HF. Несмотря на это, плотность заряда, выражаемая через 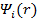 , является правильной.
, является правильной.
Для того чтобы рассчитать полную энергию и электронную плотность методом DFT, необходимо вычислить орбитали , которые являются решением уравнения. Для того, чтобы записать это уравнение необходимо знать ядерный потенциал 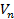 (который мы задаем при формулировке задачи), электронный потенциал
(который мы задаем при формулировке задачи), электронный потенциал 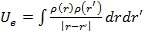 и корреляционный вклад.
и корреляционный вклад. 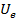 можно определить, решив уравнение Пуассона:
можно определить, решив уравнение Пуассона:
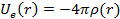 .
.
Также необходимо уравнение для вычисления корреляционного вклада. Таким образом, мы получим систему уравнений, решив которую, мы получим электронную плотность.
Поскольку система является нелинейной, основным методом ее решения является итерационный. На первом шаге каким-либо образом выбирается и фиксируется начальная электронная плотность r0(r).
После этого для нее решается система уравнений, что дает набор 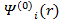 . Затем, используя (3.23), можно вычислить новую электронную плотность и повторить процесс. Эти итерации необходимо повторять до тех пор, пока
. Затем, используя (3.23), можно вычислить новую электронную плотность и повторить процесс. Эти итерации необходимо повторять до тех пор, пока 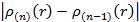 будет меньше выбранной погрешности. Полученная электронная плотность будет искомой. После этого можно вычислить полную энергию по формуле:
будет меньше выбранной погрешности. Полученная электронная плотность будет искомой. После этого можно вычислить полную энергию по формуле:
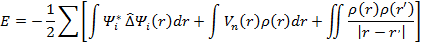
Используя вычисленные величины можно применить процедуры для вычисления различных свойств. Как было сказано выше, приближение LDA рассчитано на системы с медленно меняющейся электронной плотностью. Несмотря на это, она довольно точна и для систем с быстро меняющейся плотностью, например, твердых тел. К сожалению, для задач квантовой химии LDA оказывается недостаточно точной. К тому же она не учитывает спиновые корреляции.
Проблема учета спиновых корреляций может быть решена довольно просто привлечением приближения локальной спиновой плотности, в которой корреляционная энергия определяется как:
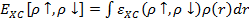 .
.
Для этого приближения значение 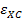 было вычислено достаточно точно с помощью стохастических методов (квантовый метод Монте-Карло) при расчетах систем свободных электронов.
было вычислено достаточно точно с помощью стохастических методов (квантовый метод Монте-Карло) при расчетах систем свободных электронов.
Дата добавления: 2015-04-19; просмотров: 3991;