Важнейшие алхимические знаки 5 страница
Теория химического строения Бутлерова, в основе которой лежит теория валентности, позволила дать объяснение многим экспериментальным фактам, касавшимся изомерии органических соединений и их реакционной способности. Учебник Бутлерова "Введение к полному изучению органической химии" (1866) уже через год – впервые в истории отечественной науки – был опубликован на немецком языке. В 1862 г. немецкий химик Эмиль Эрленмейер пришёл к идее о существовании двойной связи углерод-углерод в этилене и тройной в ацетилене. Положение Бутлерова о взаимном влиянии атомов в молекуле активно развивал и другой русский химик – Владимир Васильевич Марковников – представивший в 1869 г. диссертацию "Материалы по вопросу о взаимном влиянии атомов в химических соединениях".
Огромным достоинством теории химического строения явилась возможность наглядного изображения молекулы. История развития графического изображения валентности и химической связи сама по себе чрезвычайно интересна. Ниже приведены различные способы изображения формулы пропионовой кислоты в нескольких системах 1860-х годов.

Т.н. "роликовые" модели, предложенные Кекуле в 1857 г., не были приняты научным сообществом, и сам автор от них быстро отказался. Некоторое время многими химиками использовались формулы, напоминающие формулы теории типов Жерара-Лорана, в которых связь между атомами и радикалами обозначалась с помощью фигурных скобок (одним из первых изображать в формулах теории типов чёрточку, соединяющую символы элементов, начал в 1864 г. Лотар Мейер). Шотландский химик Александер Крум Браун предложил в 1864 г. использовать структурные формулы в виде окружностей с помещёнными в них символами элементов, соединённых линиями, обозначающими химическую связь между атомами; количество линий соответствовало валентности атома. В 1865 г. немецкий химик-органик Август Вильгельм фон Гофман продемонстрировал первые шаростержневые модели, в которых роль атомов играли крокетные шары, получившие распространение под названием "глиптических" (глиптика – искусство резьбы по камню). В 1866 г. в учебнике Кекуле появились рисунки моделей, в которых атом углерода уже имел тетраэдрическую конфигурацию.
Весьма важным для развития системы структурных формул стало установление строения молекулы бензола и, как следствие, огромного числа ароматических соединений. Замкнутое кольцо из шести атомов углерода, связанных попеременно одной или двумя валентностями предложил в 1865 г. Кекуле. Впрочем, попытки предложить формулу, объясняющую равноценность всех атомов углерода в молекуле бензола продолжались ещё долгое время.
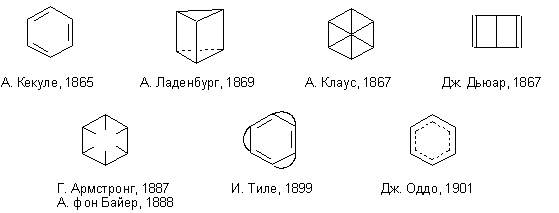
Существенный вклад в развитие классической теории валентности в применении к ненасыщенным соединениям внёс немецкий химик Иоганн Тиле, предложивший в 1899 г. теорию парциальных валентностей. Согласно этой теории, при образовании кратной связи между атомами углерода валентности этих атомов не насыщаются полностью, но каждый атом сохраняет свободной часть сродства – парциальную валентность. Если две двойные связи разделены одинарной, то образуется система сопряжённых двойных связей: парциальные валентности средних атомов углерода насыщаются, а парциальные валентности крайних остаются свободными. Теория парциальных валентностей удачно дополнила первоначальное учение о валентности, позволив дать объяснение многим реакциям, в частности, реакциям присоединения к ненасыщенным углеводородам.
Ещё одной попыткой усовершенствования теории валентности стала теория мезогидрии, которую разработал итальянский химик Джузеппе Оддо. Оддо предположил, что атом водорода, находясь вблизи двух атомов многовалентных элементов, может делить свою валентность между ними; его теория позволила дать объяснение, в частности, явлению таутомерии.
Стереохимия
Важнейшим этапом развития структурной химии стало установления пространственного строения молекул. Ещё в 1867 г. Кекуле критиковал формулы Крум-Брауна, поскольку считал, что такой вид изображения даёт неверное представление, будто все валентности находятся в одной плоскости. Сам Кекуле неоднократно высказывал предположение о тетраэдрическом расположении четырёх валентностей углерода.
Возникновение стереохимии было обусловлено целым рядом открытий, поначалу, казалось, не имевших отношения к химии. В 1801 г. Томас Юнг провёл опыты, доказавшие волновую природу света. Огюстен Жан Френель около 1814 г. показал, что световые волны относятся к типу поперечных волн, объяснив природу плоскополяризованного света, который открыл в 1808 г. Этьен Луи Малюс. В 1815 г. французский физик Жан Батист Био открыл явление оптической активности некоторых веществ. Выделенные Берцелиусом в 1832 г. изомерные винная и виноградная кислоты (винная вращает плоскость поляризации света вправо, виноградная оптически неактивна) стали первым примером оптической изомерии.
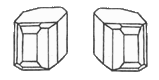
|
| Энантиоморфные кристаллы тартрата натрия-аммония |
В 1848 г. французский химик и биолог Луи Пастер обнаружил, что виноградная кислота представляет собой смесь двух оптических антиподов – винной кислоты и её изомера, отличающегося лишь тем, что он вращает плоскость поляризации света влево. Кристаллы этих изомеров оказались асимметричными; Пастер высказал предположение о том, что асимметрия кристаллов и оптическая активность должны быть обусловлена асимметрией молекул. Иоганн Адольф Вислиценус, открывший изомеры молочной кислоты, опять-таки различающиеся только оптической активностью, писал в 1873 г.: "различие оптических изомеров можно объяснить различным расположением их атомов в пространстве, и надлежит искать определённых представлений об этом расположении".
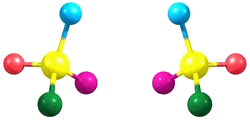
|
| Асимметричные атомы углерода, являющиеся оптическими антиподами |
Гипотезу, объясняющую оптическую изомерию – гипотезу асимметричного атома углерода – предложил в 1774 г. датский химик Якоб Генрик Вант-Гофф. Вант-Гофф основывался на предположении о том, что четыре валентности атома углерода направлены из центра тетраэдра к вершинам. Симметрия нарушается в том случае, когда все четыре атома или радикала, соединённые с данным атомом углерода, различны. Присоединение в этом случае может произойти двумя различными способами, и полученные фигуры представляют собой зеркальное отображение друг друга – как раз тот тип асимметрии, который наблюдал Пастер в кристаллах винных кислот.
Почти одновременно с Вант-Гоффом подобные предположения опубликовал французский химик Жозеф Ашиль Ле Бель. Гипотеза Вант-Гоффа – Ле Беля очень удачно предсказывала оптическую активность соединений и количество оптических изомеров. В 1875 г. в новом издании своей брошюры "Химия в пространстве" Вант-Гофф дал объяснение оптической активности соединений алленового типа, показав, что эти вещества обладают асимметричным строением и должны поэтому иметь правую и левую формы даже при отсутствии асимметричного атома углерода.
Несмотря на блестящее совпадение выводов Вант-Гоффа с опытными данными, новая теория (активно пропагандируемая Вислиценусом) вызвала ожесточённую дискуссию; Кекуле отнёсся к ней весьма скептически, а выдающийся немецкий химик-органик Адольф Вильгельм Герман Кольбе выступил в 1877 г. с чрезвычайно резкой критикой в адрес Вант-Гоффа и Вислиценуса. Тем не менее, даже авторитет Кольбе оказался не в силах затмить несомненные достоинства теории. В защиту стереохимии выступили Вильгельм Фридрих Оствальд, Шарль Адольф Вюрц, Альберт Ладенбург. В 1885 г. немецкий химик Иоганн Фридрих Вильгельм Адольф фон Байер суммировал положения классической теории валентности и стереохимии, разработав теорию напряжения, призванной объяснить устойчивость циклических и непредельных соединений, в основе которой лежит представление о тетраэдрическом углеродном атоме. Вислиценус в 1887 г. на основе теории Вант-Гоффа дал надлежащее объяснение геометрической изомерии на примере малеиновой и фумаровой кислот. Окончательное утверждение теории Вант-Гоффа состоялось в 1890-х гг. благодаря блестящим синтетическим работам, которые выполнили Пауль Вальден, открывший т.н. вальденовское обращение, и Эмиль Герман Фишер.
Координационная химия
На протяжении довольно долгого времени теория валентности применялась главным образом к органическим соединениям. Однако довольно скоро структурные представления оказались востребованы также и в химии комплексных соединений. Теоретические представления этого раздела неорганической химии формировались на основе изучения свойств комплексов, получаемых взаимодействием солей переходных металлов с аммиаком. Первым шагом на пути к координационной химии стала аммонийная гипотеза Томаса Грэма (1840 г.), усмотревшего аналогию между взаимодействием аммиака с кислотами и с солями металлов; согласно этой гипотезе, металл занимал место одного из атомов водорода в ионе аммония. Гипотеза Грэма была развита в 1851 г. Гофманом, предположившим, что атом водорода в аммонийном радикале способен замещаться на другой аммонийный радикал.
Следующим шагом стала цепная теория, предложенная в 1869 г. Кристианом Вильгельмом Бломстрандом и усовершенствованная Софусом Мадсом Йёргенсеном. В теории Бломстранда – Йёргенсена для некоторых элементов допускалась валентность выше обычной, а также возможность образования цепей атомами азота, кислорода и других элементов. Экспериментально установленное различие между кислотными остатками, входящими в состав комплекса, объяснялось различным способом их связывания – непосредственно с металлом или с концом цепи. Например, для аммиакатных комплексов состава CoCl3·6NH3, CoCl3·5NH3 и CoCl3·4NH3, из растворов которых нитратом серебра осаждаются три, два и один эквивалент хлора соответственно, Йёргенсен предполагал следующее строение:
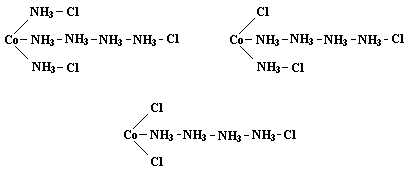
Однако теория Бломстранда – Йёргенсена не могла объяснить, например, существование двух изомерных комплексов состава CoCl3·4NH3 – празеосоли (зелёный) и виолеосоли (фиолетовый).
В 1893 г. швейцарский химик Альфред Вернер опубликовал статью "О строении неорганических соединений", в которой изложил основные положения созданной им координационной теории. Теория Вернера (который уже был известен своими работами по стереохимии органических соединений азота) существенно расширила представления химиков о валентности элементов.
Главное положение теории состояло в признании за атомами способности, наряду со связями сродства (главная, первичная или обычная валентность), образовывать дополнительные (координационные) связи (побочная или вторичная валентность). Вернер ввёл понятие о комплексном ионе – сложной комбинации атомов и молекул, состоящем из центрального иона, связанного с несколькими молекулами или отрицательными ионами (лигандами), образующими внутреннюю сферу комплекса; главной характеристикой центрального иона Вернер считал координационное число, равное сумме главной и побочной валентностей. Для упоминавшихся выше комплексных солей кобальта в рамках координационной теории предполагалось строение с координационным числом 6:
[Co(NH3)6]Cl3 [Co(NH3)5Cl]Cl2 [Co(NH3)4Cl2]Cl
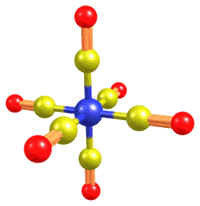
|
| Октаэдрическая молекула карбонила хрома Cr(CO)6 |
Важной частью координационной теории стали положения, касающиеся стереохимии комплексных соединений. Вернер предположил, что для комплексов с координационным числом 6 валентности направлены к вершинам октаэдра, в центре которого находится центральный ион; для комплексов с координационным числом 4 валентности направлены либо к вершинам тетраэдра, либо к углам квадрата. В связи с этими положениями Вернер подробно рассмотрел возможные случаи изомерии соответствующих комплексов, в частности, цис-транс-изомерии в квадратных и октаэдрических комплексах и оптической изомерии в тетраэдрических и октаэдрических комплексах.
Теория Вернера наглядно объяснила строение комплексных соединений, легла в основу их классификации и позволила предсказать существование большого числа новых соединений. Слабой стороной теории являлись положения о двух типах валентности, которые невозможно было обосновать теоретически. Это сделало ещё более острой главную проблему структурной химии XIX века (да и всей атомно-молекулярной теории): после отказа от электрохимического дуализма Берцелиуса в распоряжении учёных не имелось хоть сколько-нибудь адекватных представлений о природе валентных сил. Блестящие успехи химической теории, казалось, нисколько не приблизили естествоиспытателей к ответу на вопрос о способе соединения атомов. Как и в случае периодического закона, ответ оказался за пределами предмета химии.
5.3. УЧЕНИЕ О ХИМИЧЕСКОМ ПРОЦЕССЕ – ФИЗИЧЕСКАЯ ХИМИЯ
Термохимия - Термодинамика - Химическое равновесие - Химическая кинетика - Катализ - Учение о растворах
Граница между химией и физикой уже в момент появления научной химии была весьма условной. На протяжении XVII – XVIII веков ещё можно было говорить о более или менее явных различиях в предметах этих наук; к предмету химии относились процессы, сопровождавшиеся изменением состава вещества. Однако начало XIX века ознаменовалось стремительным взаимным проникновением физики и химии. Атомно-молекулярную теорию вряд ли можно счесть исключительно химической теорией; изучение тепловых эффектов, сопровождающих химические реакции, нельзя считать предметом одной только химии. Создание гальванического элемента, электролиз, открытие оптической активности веществ, установление связи между теплоёмкостью и атомной массой существенно размыли границу между этими науками.
К середине XIX века окончательно оформилась и начала стремительно развиваться пограничная область науки – физическая химия. Предметом изучения новой науки можно считать химический процесс – его скорость, направление, сопровождающие его тепловые явления и зависимость этих характеристик от внешних условий. Законы, описывающие химический процесс, могут иметь одинаковый вид для совершенно различных процессов.
Термохимия
Уже в конце XVIII века появились первые работы, посвящённые изучению тепловых эффектов химических реакций, с которых и началось становление термохимии. Выделение теплоты как одна из составляющих химического процесса рассматривалось уже во флогистонной теории. Следует отметить, что термохимия сразу же приобрела важное практическое значение: определение теплоты горения различных видов топлива имело большое значение для техники. Однако уже в первой половине XIX века химики рассматривали термохимию ещё и как важный инструмент для решения таких важнейших теоретических проблем химии, как изучение состава и строения вещества и определения сил химического сродства. Теплоту начали рассматривать и как возможную меру прочности соединения атомов и взаимодействия молекул.
Следует отметить, что во время возникновения термохимии взгляды на природу теплоты существенно отличались от современных. Естествоиспытатели исходили из предположения, что существует некая невесомая материальная субстанция – теплород – высвобождающаяся либо поглощающаяся в ходе химической реакции.
Иное представление о теплоте как о форме движения мельчайших частиц материи (атомов) появилось еще в XVII веке. Этих воззрений придерживались Фрэнсис Бэкон, Рене Декарт, Исаак Ньютон, Роберт Гук. Швейцарский математик и физик Даниил Бернулли предложил в 1734 г. уравнение, связывающее давление газа с движением атомов. Михаил Васильевич Ломоносов, развивая свою корпускулярную философию, детально разработал положения кинетической теории теплоты. Однако без доказательства существования атомов кинетические воззрения на природу теплоты не могли найти всеобщего признания. В 1798 г. Бенджамин Томпсон (граф Румфорд), описав выделение большого количества тепла при высверливании канала в пушечном стволе, посчитал это экспериментальным доказательством того, что теплота является формой движения. Конечно, факт выделения тепла при трении был известен с незапамятных времен; однако сторонники теории теплорода усматривали в этом явлении нечто аналогичное электризации тел трением – трение способствует выжиманию теплорода из тела. Румфорд, однако, показал, что из ограниченного количества материи может быть получено неограниченное количество теплоты. Получение теплоты с помощью трения подтвердили и опыты Гемфри Дэви, обнаружившего, что трение двух кусков льда друг о друга вызывает их плавление. После утверждения атомно-молекулярной теории теплородная теория теплоты в значительной степени поколебалась, однако в первой половине XIX века концепция теплорода разделялась большинством ученых.
Одним из первых систематическое изучение тепловых явлений начал английский химик Джозеф Блэк. Он сформулировал понятия теплоёмкости тел и скрытой теплоты изменения агрегатного состояния вещества, указал на необходимость чёткого различения между количеством и интенсивностью теплоты (температурой). С помощью изобретённого им калориметра Блэк в 1759-1763 гг. выполнил первые определения теплоёмкостей веществ и скрытой теплоты плавления льда и испарения воды.
Первые систематические опыты по измерению теплот химических реакций начали в 1780 г. французские химики Антуан Лоран Лавуазье и Пьер Симон де Лаплас. Обобщая результаты своих исследований, Лавуазье и Лаплас сформулировали правило, известное сейчас как первый закон термохимии (закон Лавуазье-Лапласа): "Если при соединении или при любом изменении состояния системы происходит уменьшение свободной теплоты, то эта теплота в полной мере появится вновь, когда вещества вернутся к своему первоначальному состоянию, и наоборот…".
Важнейшую роль в развитии термохимии сыграл русский химик Герман Иванович Гесс. В период с 1830 по 1850 гг. им был проведён ряд систематических исследований в области термохимии. В 1840 г. Гесс сформулировал фундаментальный закон термохимии – закон постоянства количества теплоты: "Каким бы путём не совершалось соединение, – имело ли оно место непосредственно или происходило косвенным путём в несколько приёмов, – количество выделившейся при его образовании теплоты всегда постоянно". Гесс не только открыл основной закон термохимии, доказав его экспериментально, но и широко применял этот закон для расчётов теплот различных процессов, которые невозможно определить непосредственно. Закон Гесса выражает принцип сохранения энергии применительно к химическим процессам, являясь следствием первого начала термодинамики. Гесс одним из первых высказал предположение, что определение теплоты реакции может дать возможность измерить химическое сродство; впоследствии эта идея легла в основу принципа максимальной работы Бертло-Томсена.
В 60-е годы XIX века независимо друг от друга два выдающихся исследователя – Пьер Эжен Марселен Бертло и Ханс Петер Юрген Юлиус Томсен высказали предположение о том, что тепловой эффект реакции является мерой химического сродства между реагентами. Основное положение данного подхода Бертло сформулировал следующим образом: "Теплота, выделяющаяся при реакции, служит мерой суммы физических и химических работ, совершаемых при этой реакции. …Принцип максимальной работы заключается в том, что всякое <самопроизвольное> химическое превращение… стремится к образованию тела или системы тел, которые выделяют наибольшее количество тепла". Считая, что реакции самопроизвольно протекают лишь тогда, когда сопровождаются выделением теплоты, Бертло многие годы посвятил систематическому определению тепловых эффектов химических реакций. По его мнению, результаты термохимических измерений должны были позволить вычислять направление реакции и предвидеть принципиальную осуществимость химического взаимодействия.
Следует отметить, что принцип максимальной работы Бертло-Томсена критиковался многими современниками, поскольку далеко не все самопроизвольные реакции сопровождаются выделением теплоты; кроме того, уже были известны обратимые реакции. Тем не менее, хотя принцип максимальной работы не является общим законом природы, в ряде случаев (особенно в области низких температур) его можно было успешно использовать для предсказания направления реакции.
Термодинамика
Важнейшую роль в создании представлений о химическом сродстве и химическом процессе сыграли физические исследования середины XIX века в области термодинамики. Еще в 1765 г. Джеймс Уатт начал экспериментальные исследования паровой машины, которые затем были продолжены широким кругом исследователей. Никола Леонар Сади Карно, исследуя практическую задачу получения работы из тепла применительно к паровым машинам, предложил рассматривать этот принцип в самом общем смысле, сформулировав тем самым общий метод решения задачи – термодинамический, заложивший основу термодинамики. Определяя коэффициент полезного действия тепловых машин, Карно вывел свой знаменитый цикл, КПД которого не зависит от свойств рабочего тела (пара, газа и т.д.) и определяется лишь температурами теплоотдатчика и теплоприемника.
Карно первым вскрыл связь теплоты с работой, хотя и исходил поначалу из концепции теплорода, признававшей теплоту неизменной по количеству субстанцией. Так или иначе, Карно заложил основы термодинамики как раздела физики, изучающего наиболее общие свойства макроскопических систем, находящихся в состоянии термодинамического равновесия, и процессы перехода между этими состояниями. Термодинамика стала развиваться на основе фундаментальных принципов или начал, являющихся обобщением результатов многочисленных наблюдений и экспериментов.
Первое начало термодинамики (закон сохранения энергии в применении к термодинамическим процессам) гласит: при сообщении термодинамической системе определенного количества теплоты в общем случае происходит приращение внутренней энергии системы, и она совершает работу против внешних сил. Выше отмечалось, что первым, кто поставил теплоту в связь с работой, был Карно, но его работа о механическом эквиваленте теплоты в силу запоздалой публикации не оказала решающего воздействия на формирование первого начала термодинамики. Однако идея о том, что теплота – не субстанция, а сила (энергия), одной из форм которой и является теплота, причем эта сила, в зависимости от условий, выступает в виде движения, электричества, света, магнетизма, теплоты, которые могут превращаться друг в друга, существовала в умах исследователей. Для превращения этой идеи в ясное и точное понятие необходимо было определить общую меру этой силы. Это сделали в 40-х годах XIX века независимо друг от друга Р. Майер, Дж. Джоуль и Г. Гельмгольц.
Юлиус Роберт Майер в 1842 г. сформулировал закон эквивалентности механической работы и теплоты и рассчитал механический эквивалент теплоты. Джеймс Прескотт Джоуль экспериментально подтвердил предположение о том, что теплота является формой энергии, и экспериментально доказал эквивалентность превращения механической работы в теплоту. Герман Людвиг Фердинанд Гельмгольц в 1847 г. математически обосновал закон сохранения энергии, показав его всеобщий характер.
Рудольф Юлиус Иммануил Клаузиус и Уильям Томсон (лорд Кельвин) в начале 50-х годов XIX века сформулировали второе начало термодинамики, утверждающее невозможность самопроизвольного перехода теплоты от менее нагретого тела к более нагретому (Клаузиус) и невозможность полного преобразования теплоты в работу (Томсон). Клаузиус начал детально разрабатывать механическую теорию теплоты, ввёл в термодинамику важнейшие понятия внутренней энергии и энтропии.
Распространив принцип возрастания энтропии замкнутой системы на всю Вселенную, Клаузиус высказал гипотезу о тепловой смерти Вселенной. Концепция "тепловой смерти" у Клаузиуса заключена в формулировке второго начало термодинамики в виде следующего положения: "Энтропия Вселенной стремится к максимуму". Постулат Клаузиуса и концепция тепловой смерти вызвали большое количество возражений; были придуманы многочисленные эксперименты, казалось, противоречащие принципу Клаузиуса. Очень тонкий мысленный эксперимент, например, выдвинул в 1870 г. Максвелл (т.н. "демон Максвелла").
Развивая взгляды Клаузиуса, Людвиг Больцман и Джеймс Клерк Максвелл в 70-е годы XIX в. показали статистический характер второго начала термодинамики. Формулировки второго начала термодинамики не соответствовали традиционным механическим представлениям, согласно которым все процессы обратимы. Кинетическая теория теплоты сделала это несоответствие противоречием. Эти трудности были преодолены Максвеллом и Больцманом, которые ввели понятие вероятности физических явлений и поставили на место динамических законов в механике статистические законы в термодинамике. Закон возрастания энтропии у Больцмана получил простую интерпретацию: "Система стремится к наиболее вероятному состоянию".
Значительно позже – в 1906-1911 гг. – различные формулировки третьего начала термодинамики, позволившего рассчитывать абсолютные значения энтропии вещества, предложили Вальтер Герман Нернст и развивший его взгляды Макс Карл Эрнст Людвиг Планк.
Окончательно взаимосвязь между механическими процессами и тепловыми явлениями была установлена в кинетической теории газов. Гельмгольц в 1847 г. выдвинул гипотезу о том, что внутренняя причина взаимопревращения механической работы и теплоты лежит в сведении тепловых явлений к явлениям механического движения. В 1856-57 г. немецкий физик Август Карл Кренинг и Рудольф Клаузиус разработали кинетическую теорию теплоты, получив уравнение, связывающее среднюю кинетическую энергию молекул с температурой. Статистический метод в теорию газового состояния ввели в 1860-1865 гг. Л. Больцман и Дж. Максвелл. Большое значение для создания кинетической теории теплоты имели исследования законов газового состояния. На основе законов Бойля и Гей-Люссака французский физик Бенуа Поль Эмиль Клапейрон получил в 1834 г. уравнение состояния идеального газа (уравнение Клапейрона), обобщённое в 1874 г. Д.И. Менделеевым. В 1871 г. в своей дипломной работе Иоганн Дидерик Ван-дер-Ваальс вывел уравнение состояния реальных газов, учитывающее взаимное притяжение молекул газа и их собственный объём.
Химическое равновесие
В результате соединения классической термодинамики и термохимии появился важнейший раздел физической химии – химическая термодинамика. Одной из первоочередных задач химической термодинамики стало изучение химического равновесия. В середине XIX века появились экспериментальные данные, возродившие интерес к идеям Бертолле, в частности, к введённому им понятию химической массы, которое, по мнению Бертолле, определяет протекание реакции и состав её продуктов. В 1850 г. Александер Уильям Уильямсон, исследуя сложные эфиры, установил, что реакции этерификации являются обратимыми и приводят к наступлению динамического равновесия, в котором присутствуют и исходные вещества, и продукты реакции. Немецкие химики Генрих Розе и Роберт Вильгельм Бунзен в 1851-1853 гг. показали, что реакции обмена часто являются обратимыми и направление реакции можно изменить путём подбора соответствующих условий её протекания. В 1857 г. Анри Этьенн Сент-Клер Девиль опубликовал данные о термической диссоциации веществ, в частности, о диссоциации воды на водород и кислород; в 1859 г. Николай Николаевич Бекетов начал серию работ по изучению зависимости от внешних условий явления вытеснения одним элементом другого из его соединений. Именно эти исследования и положили начало химической динамике: наряду с изучением состава и строения соединений (химической статикой, по выражению Бекетова) химики начали изучение закономерностей протекания химических процессов.
В 1862-1867 гг. Марселен Бертло и Анри Дебре сделали первые обобщения о зависимости предела протекания обратимых реакций от количеств исходных веществ и давления газообразных продуктов реакции. Наконец, в 1864-1867 гг. норвежские учёные Като Максимилиан Гульдберг и Петер Вааге опубликовали серию работ, в которой излагался закон действующих масс. Представляя равновесие обратимой реакции как равенство двух сил сродства, действующих в противоположных направлениях, Гульдберг и Вааге показали, что направление реакции определяется не массами веществ (как у Бертолле), а произведением действующих масс (концентраций) реагирующих веществ. Закон действующих масс, гласивший, что отношение произведений действующих масс исходных веществ и продуктов реакции есть величина постоянная (константа равновесия), представлял собой общий путь вычисления условий равновесия для любых количеств реагирующих веществ. Однако работа Гульдберга и Вааге "Исследование сил химического сродства" (1867 г.) была переведена на немецкий язык лишь в 1879 г. и только после этого приобрела известность в научном сообществе. После 1880 г. закон действующих масс стали рассматривать как один из основополагающих законов химии.
В 1874-1878 гг. американский физик Джозайя Уиллард Гиббс опубликовал серию работ, посвящённых теоретическому рассмотрению термодинамики химического равновесия; Гиббс ввёл в термодинамику понятия свободной энергии, термодинамического и химического потенциалов. Исследования Гиббса составили фундамент современной химической термодинамики. К сожалению работы Гиббса также стали известны с большим опозданием, поскольку были опубликованы в "Трудах Коннектикутской Академии наук" – издании, совершенно не знакомом европейскому научному миру. Вследствие этого многие выводы Гиббса были впоследствии получены независимо от него другими исследователями.
Дата добавления: 2015-04-15; просмотров: 1854;